The Impact of Oxalate Ions on Barium Sulfate Crystallization
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Crown Chemical Resistance Chart
Crown Polymers, Corp. 11111 Kiley Drive Huntley, IL. 60142 USA www.crownpolymers.com 847-659-0300 phone 847-659-0310 facisimile 888-732-1270 toll free Chemical Resistance Chart Crown Polymers Floor and Secondary Containment Systems Products: CrownShield covers the following five (4) formulas: CrownShield 50, Product No. 320 CrownCote, Product No. 401 CrownShield 40-2, Product No. 323 CrownShield 28, Product No. 322 CrownPro AcidShield, Product No. 350 CrownCote AcidShield, Product No. 430 CrownPro SolventShield, Product No. 351 CrownCote SolventShield, Product No. 440 This chart shows chemical resistance of Crown Polymers foundational floor and secondary containment product line that would be exposed to chemical spill or immersion conditions. The chart was designed to provide general product information. For specific applications, contact your local Crown Polymers Floor and Secondary Containment Representative or call direct to the factory. ; Resistant to chemical immersion up to 7 days followed by wash down with water 6 Spillage environments that will be cleaned up within 72 hours after initial exposure. 9 Not Recommended Chemical CrownShield SolventShield AcidShield Chemical CrownShield SolventShield AcidShield 1, 4-Dichloro-2-butene 9 6 6 Aluminum Bromate ; ; ; 1, 4-Dioxane 9 6 6 Aluminum Bromide ; ; ; 1-1-1 Trichloroethane 9 ; ; Aluminum Chloride ; ; ; 2, 4-Pentanedione 6 ; 6 Aluminum Fluoride (25%) ; ; ; 3, 4-Dichloro-1-butene 6 6 6 Aluminum Hydroxide ; ; ; 4-Picoline (0-50%) 9 6 6 Aluminum Iodine ; ; ; Acetic Acid (0-15%) 9 6 6 -

Optical Absorption in Gel Grown Barium Oxalate Single Crystals
OPTOELECTRONICS AND ADVANCED MATERIALS – RAPID COMMUNICATIONS Vol. 4, No. 11, November 2010, p. 1713 - 1716 Optical absorption in gel grown barium oxalate single crystals P. V. DALAL*, K. B. SARAF, M. P. DESHPANDEa P. G. Department of Physics, Pratap College, Amalner -425 401 aDepartment of Physics, Sardar Patel University, Vallabh Vidyanagar -388 120, Gujarat, India Single crystals of barium oxalate have been grown by simple gel technique using agar gel as the growth medium at ambient temperature. The slow and controlled reaction between barium chloride and oxalic acid in agar gel has formed barium oxalate. Optical absorption spectra of this grown crystal is recorded in the wavelength region from 200 to 800 nm. The absorption spectra reveal transitions involving absorption and emission of phonons and also show that the crystal is transparent in the region 500 to 800 nm. The detail study supports the existence of forbidden indirect transition in the material. Different segment of α1/3 vs hν graph were used to distinguish individual contribution of phonons and scattering of charge carriers in the lattice is found due to acoustic phonons. (Received October 27, 2010; accepted November 10, 2010) Keywords: Barium oxalate single crystal, Agar-agar gel, X-ray diffraction, Photo-absorption, Photon energy 1. Introduction The reaction, which leads to the growth of crystals can be, expressed as Barium oxalate is a pyro-nature material that shows great promise in pyrotechnic and high temperature BaCl2 + H2C2O4 = BaC2O4 +2HCl electronic applications. The high dielectric constant and melting point of barium oxalate is an advantage to The optimum conditions for the growth of barium improve hardness of barium titanate in capacitor industries oxalate single crystals were: concentration of gel 1.5%, [1]. -

Chemical Resistance 100% SOLIDS EPOXY SYSTEMS
Chemical Resistance 100% SOLIDS EPOXY SYSTEMS CHEMICAL 8300 SYSTEM 8200 SYSTEM 8000 SYSTEM OVERKOTE PLUS HD OVERKOTE HD OVERKRETE HD BASED ON ONE YEAR IMMERSION TESTING –––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––––– Acetic Acid (0-15%) G II Acetonitrile LLG L Continuous Immersion Acetone (0-20%) LLL Acetone (20-30%) Suitable for continuous immersion in that chemical (based on LLG Acetone (30-50%) L G I ONE YEAR testing) to assure unlimited service life. Acetone (50-100%) G II Acrylamide (0-50%) LLL G Short-Term Exposure Adipic Acid Solution LLL Alcohol, Isopropyl LLL Suitable for short-term exposure to that chemical such as Alcohol, Ethyl LLG secondary containment (72 hours) or splash and spill Alcohol, Methyl LLI (immediate clean-up). Allyl Chloride LLI Allylamine (0-20%) L L I Allylamine (20-30%) L G I I Not Suitable Allylamine (30-50%) GGI Not suitable for any exposure to that chemical. Aluminum Bromide LL– Aluminum Chloride L L – Aluminum Fluoride (0-25%) L L – This chart shows chemical resistance of our various Aluminum Hydroxide LLL 1 topping materials (90 mils – ⁄4"). These ratings are based on Aluminum Iodide LL– temperatures being ambient. At higher temperatures, chemical Aluminum Nitrate LL– resistance may be effected. When chemical exposure is Aluminum Sodium Chloride L L – minimal to non-existent, a 9000 System–FlorClad™ HD or Aluminum Sulfate LLL 4600 System– BriteCast™ HD may be used. Alums L L L 2-Aminoethoxyethanol Resistance data is listed with the assumption that the material GGG has properly cured for at least four days, at recommended Ammonia – Wet L L – temperatures, prior to any chemical exposure. -

High Purity Inorganics
High Purity Inorganics www.alfa.com INCLUDING: • Puratronic® High Purity Inorganics • Ultra Dry Anhydrous Materials • REacton® Rare Earth Products www.alfa.com Where Science Meets Service High Purity Inorganics from Alfa Aesar Known worldwide as a leading manufacturer of high purity inorganic compounds, Alfa Aesar produces thousands of distinct materials to exacting standards for research, development and production applications. Custom production and packaging services are part of our regular offering. Our brands are recognized for purity and quality and are backed up by technical and sales teams dedicated to providing the best service. This catalog contains only a selection of our wide range of high purity inorganic materials. Many more products from our full range of over 46,000 items are available in our main catalog or online at www.alfa.com. APPLICATION FOR INORGANICS High Purity Products for Crystal Growth Typically, materials are manufactured to 99.995+% purity levels (metals basis). All materials are manufactured to have suitably low chloride, nitrate, sulfate and water content. Products include: • Lutetium(III) oxide • Niobium(V) oxide • Potassium carbonate • Sodium fluoride • Thulium(III) oxide • Tungsten(VI) oxide About Us GLOBAL INVENTORY The majority of our high purity inorganic compounds and related products are available in research and development quantities from stock. We also supply most products from stock in semi-bulk or bulk quantities. Many are in regular production and are available in bulk for next day shipment. Our experience in manufacturing, sourcing and handling a wide range of products enables us to respond quickly and efficiently to your needs. CUSTOM SYNTHESIS We offer flexible custom manufacturing services with the assurance of quality and confidentiality. -

Studies on the Growth and Characterization of Barium Doped Copper Cadmium Oxalate Dihydrate Single Crystals
Journal of Materials and J. Mater. Environ. Sci., 2020, Volume 11, Issue 5, Page 788-794 Environmental Science ISSN : 2028-2508 CODEN : JMESCN http://www.jmaterenvironsci.com Copyright © 2020, University of Mohammed Premier Oujda Morocco Studies on the Growth and Characterization of Barium Doped Copper Cadmium Oxalate Dihydrate Single Crystals P. S. Rohith1, N. Jagannatha1 *, K.V. Pradeepkumar1 1 PG Department of Physics, FMKMC College, A Constituent College of Mangalore University, Madikeri-571201, Karnataka, India Received 22 Feb 2020, Abstract Revised 28 April 2020, Single crystals Barium doped copper cadmium oxalate (BCuCO) was grown from the Accepted 28 April 2020 silica hydrogel using a single diffusion technique. The grown crystals were characterized using energy-dispersive X-ray spectroscopy (EDX), single-crystal X-ray Diffraction Keywords studies, powdered X- Ray Diffraction, FTIR spectroscopy, UV-visible BCuCO, Spectrophotometer, and thermal studies. Energy-dispersive X-ray spectroscopy (EDX) Single diffusion confirmed the presence of Ba, Cd, and Cu elements in the lattice of BCuCO crystal. Triclinic, Single-crystal XRD reveals that the as-grown crystal belongs to the triclinic crystal EDX, system with the space group P . The FTIR spectroscopic studies confirmed the oxalate 1 FTIR , group, water molecule, formation of metal-oxygen bonding in BCuCO crystals. The UV- TGA. visible spectral studies reveal that the crystal is an insulator with wide bandgap and optical transparency in the visible region. The thermal stability of grown crystals, crystalline jagannathnettar@mangalor water molecules, and decomposition nature was determined using thermogravimetric euniversity.ac.in ; Phone: 9448903732; analysis (TGA). 1. Introduction Crystals are considered to possess distinct or diverse applications due to which the crystals are of higher demand and hence crystal growth is the field that is swiftly growing in the research. -

Growth and Characterization of a Novel Crystal-Barium Malate Trihydrate
IOSR Journal of Applied Physics (IOSR-JAP) e-ISSN: 2278-4861.Volume 6, Issue 3 Ver. I (May-Jun. 2014), PP 56-61 www.iosrjournals.org Growth and characterization of a novel crystal-barium malate trihydrate A Lincy, V Mahalakshmi, J Thomas and KV Saban Smart Materials Analytic Research and Technology (SMART), St. Berchmans College, Changanassery-686101, Kerala, India. Abstract: Barium malate trihydrate crystals are grown by limited diffusion technique in hydro-silica gel. Relevant functional groups are identified from the FT-IR spectrum. Single crystal XRD shows that the crystal system is monoclinic with space group P21/n. The optical band gap of the material, estimated using DRS, is 1.028 eV. The material exhibits a three stage thermal decomposition pattern. Keywords: Crystal growth, Crystal structure, Diffuse reflectance spectroscopy (DRS), Infrared spectroscopy, Thermogravimetric analysis (TGA) I. Introduction Many researchers show keen interest in the growth and characterization of metal coordinated carboxyles mainly due to their technologically important properties like ferroelectricity, magnetism and optical nonlinearity [1-3].The carboxylate ligand can coordinate with various metals leading to the formation of two and three dimensional complexes with an extensive network of hydrogen bonds [4].These hydrogen bonds between the adjacent molecules of such compounds open up a distinctive pathway for magnetic interaction among paramagnetic centers and also enhance optical non-linearity [5-6].The coordination of malic acid with various metals has resulted in many crystals which show antiferromagnetism, ferromagnetism, ferrimagnetism and optical nonlinearity [7-8]. However, metal carboxylates are only sparingly soluble in water and hence it is not easy to grow their crystals through the slow cooling and slow evaporation methods. -

Chemical Names and CAS Numbers Final
Chemical Abstract Chemical Formula Chemical Name Service (CAS) Number C3H8O 1‐propanol C4H7BrO2 2‐bromobutyric acid 80‐58‐0 GeH3COOH 2‐germaacetic acid C4H10 2‐methylpropane 75‐28‐5 C3H8O 2‐propanol 67‐63‐0 C6H10O3 4‐acetylbutyric acid 448671 C4H7BrO2 4‐bromobutyric acid 2623‐87‐2 CH3CHO acetaldehyde CH3CONH2 acetamide C8H9NO2 acetaminophen 103‐90‐2 − C2H3O2 acetate ion − CH3COO acetate ion C2H4O2 acetic acid 64‐19‐7 CH3COOH acetic acid (CH3)2CO acetone CH3COCl acetyl chloride C2H2 acetylene 74‐86‐2 HCCH acetylene C9H8O4 acetylsalicylic acid 50‐78‐2 H2C(CH)CN acrylonitrile C3H7NO2 Ala C3H7NO2 alanine 56‐41‐7 NaAlSi3O3 albite AlSb aluminium antimonide 25152‐52‐7 AlAs aluminium arsenide 22831‐42‐1 AlBO2 aluminium borate 61279‐70‐7 AlBO aluminium boron oxide 12041‐48‐4 AlBr3 aluminium bromide 7727‐15‐3 AlBr3•6H2O aluminium bromide hexahydrate 2149397 AlCl4Cs aluminium caesium tetrachloride 17992‐03‐9 AlCl3 aluminium chloride (anhydrous) 7446‐70‐0 AlCl3•6H2O aluminium chloride hexahydrate 7784‐13‐6 AlClO aluminium chloride oxide 13596‐11‐7 AlB2 aluminium diboride 12041‐50‐8 AlF2 aluminium difluoride 13569‐23‐8 AlF2O aluminium difluoride oxide 38344‐66‐0 AlB12 aluminium dodecaboride 12041‐54‐2 Al2F6 aluminium fluoride 17949‐86‐9 AlF3 aluminium fluoride 7784‐18‐1 Al(CHO2)3 aluminium formate 7360‐53‐4 1 of 75 Chemical Abstract Chemical Formula Chemical Name Service (CAS) Number Al(OH)3 aluminium hydroxide 21645‐51‐2 Al2I6 aluminium iodide 18898‐35‐6 AlI3 aluminium iodide 7784‐23‐8 AlBr aluminium monobromide 22359‐97‐3 AlCl aluminium monochloride -

Growth Aspects of Barium Oxalate Monohydrate Single Crystals in Gel Medium
Cryst. Res. Technol. 43, No. 12, 1307 – 1313 (2008) / DOI 10.1002/crat.200800038 Growth aspects of barium oxalate monohydrate single crystals in gel medium A. Moses Ezhil Raj*1, D. Deva Jayanthi2, V. Bena Jothy2, M. Jayachandran3, and 4 C. Sanjeeviraja 1 Department of Physics, Scott Christian College (Autonomous), Nagercoil-629 003, India 2 Department of Physics, Women’s Christian College, Nagercoil-629 001, India 3 ECMS Division, Central Electrochemical Research Institute, Karaikudi-630 006, India 4 Department of Physics, Alagappa University, Karaikudi-630 003, India Received 16 March 2008, revised 30 May 2008, accepted 9 June 2008 Published online 4 July 2008 Key words crystal growth, barium oxalate monohydrate, inorganic compounds, nucleation. PACS 81.10.-h, 81.10.Dn, 81.10.Aj Single crystals of barium oxalate monohydrate (BaC2O4.H2O, BOM) were grown in pure form by controlled diffusion of Ba2+ using the gel technique at different temperatures. Starting from aqueous Ba2+ chloride (BaCl2) and acetic acid (C2H2O4) in gel, this method offers a low-cost and an easiest alternative to other preparation methods for the production of barium oxalate bulky single crystals. The optimal conditions for the growth of BOM crystals in silica gel were found by investigating different growth parameters such as gel pH, gel aging and crystallization temperature. Irrespective of all such crystallization environments, growth rate of the crystals were initially less and then exhibited supersaturation effect leading to non-linearity. Gel aging and temperature has profound effect on nucleation density that resulted less number of crystals of maximum size in the gel matrix. Perfect single crystals were grown on gels of higher pH. -

Growth and Thermal Studies of Mixed Crystals of Ca-Ba Tartrate in Silica Gel
Available online a t www.scholarsresearchlibrary.com Scholars Research Library Archives of Applied Science Research, 2011, 3 (2):404-413 (http://scholarsresearchlibrary.com/archive.html) ISSN 0975-508X CODEN (USA) AASRC9 Growth and Thermal Studies of Mixed Crystals of Ca-Ba Tartrate in Silica Gel D.K. Sawant 1* , H. M. Patil 2, D.S. Bhavsar 3, J.H. Patil 4 and K D Girase 5 1-2Department of Physics, JES’s Arts, Science and Commerce College, Nandurbar (MS), India 3Department of Physics, Pratap College, Amalner (MS), India 4Department of Electronics and Telecommunication, PSGVP Mandals College of Engineering, Shahada (MS), India 5S.V.S’S Arts and Science college, Dondaicha. (MS), India ______________________________________________________________________________ ABSTRACT Mixed crystals of Calcium-Barium Tartrate were grown by a single diffusion method. The optimum conditions were established by varying various parameters such as pH of gel solution, gel concentrations, gel setting time, concentration of reactants etc. Crystals having different morphologies were obtained .Whitish semitransparent, pale yellow, rhombohedral shaped, needle shaped crystals of Calcium-Barium Tartrate were obtained. Some of them were transparent diamond shaped, some are twined. Maximum sizes of the grown crystals are 5mm×3mm and thickness about 2to3mm. The crystals grown were characterized by Thermogravimetry (TGA), Differential thermal analysis (DTA) and Derivative thermogravity (DTG). The results of these observations are described and discussed. Keywords: Gel technique, mixed tartrate crystals, TGA, DTA and DTG. ______________________________________________________________________________ INTRODUCTION Crystals are the unknown pillars of modern technology. The modern technological developments depend greatly on the availability of suitable single crystals, whether it is for lasers, semiconductors, magnetic devices, optical devices, superconductors, telecommunication etc. -

Chemical Resistance Chart
Polyurethane Thermal-Chem Corp. High Performance Epoxy Topcoats Products Chemical Resistance Chart E56 E = Excellent G = Good F = Fair Clear surfacer Plus 3.0 DecoTop < = Occasional Spillage U = Do Not Use AcidGard VersiGard ArmorTred Resurfacer Resurfacer ArmorClad Resurfacer Re DecoThane ArmorBond SolventGard DecoTop P92 Chemical 750 755 757 728 731 736 705 748 1056 1060 Polyurethane Thermal-Chem Corp. High Performance Epoxy Topcoats Products Chemical Resistance Chart E = Excellent G = Good F = Fair Clear surfacer Plus 3.0 DecoTop < = Occasional Spillage U = Do Not Use AcidGard VersiGard ArmorTred Resurfacer Resurfacer ArmorClad Resurfacer Re ThermalCrete ArmorBond SolventGard DecoFinish P81 Chemical 750 755 757 728 731 736 705 748 1030 1062 1, 4-Dichloro-2-butene U E E U U U U U U U 1, 4-Dioxane U < < U U U U U U U 1-1-1 Trichloroethane E E E F F G G F G G < < < < < 2, 4-Pentanedione F E E F F 3, 4-Dichloro-1-butene < E E U U U U U U U 4-Picoline (0-50%) U < < U U U U U V V Acetic Acid (0-5%) E E E G G G G G E G < < < < < < Acetic Acid (5-15%) E E E F Acetone(0-20%) E E E F G G G G E F Acetone (20-30% < E E < < < < < E < Acetone (30-50%) < E E < < < < < E < Acetone (50-100%) < F F < < < < < E < Acetonitrile F E E < < < < < E < Acrylamide (0-50%) E E E F G G G G F Adipic Acid Solution E E E E E E E E G Alcohol, Ethyl E E E F F F F F F Alcohol, Isopropyl E E E F F F F F G F Alcohol, Methyl < E E < < < < < < < Allyl Chloride < E E U U U U U E < Allylamine (0-20%) < E E U U U U U < Allylamine (20-30%) < E E U U U U U < Allylamine (30-50%) U < < U U U U U U Aluminum Bromate E E E F G G G G E Aluminum Bromide E E E E E E E E E Aluminum Chloride E E E E E E E E E Aluminum Fluoride (25%) E E E E E E E E E Polyurethane Thermal-Chem Corp. -
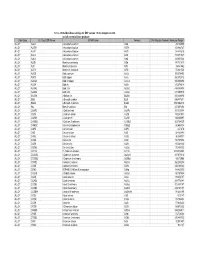
Database Full Listing
16-Nov-06 OLI Data Base Listings for ESP version 7.0.46, Analyzers 2.0.46 and all current alliance products Data Base OLI Tag (ESP) Name IUPAC Name Formula CAS Registry Number Molecular Weight ALLOY AL2U 2-Aluminum uranium Al2U 291.98999 ALLOY AL3TH 3-Aluminum thorium Al3Th 312.982727 ALLOY AL3TI 3-Aluminum titanium Al3Ti 128.824615 ALLOY AL3U 3-Aluminum uranium Al3U 318.971527 ALLOY AL4U 4-Aluminum uranium Al4U 345.953064 ALLOY ALSB Aluminum antimony AlSb 148.731537 ALLOY ALTI Aluminum titanium AlTi 74.861542 ALLOY ALTI3 Aluminum 3-titanium AlTi3 170.621536 ALLOY AUCD Gold cadmium AuCd 309.376495 ALLOY AUCU Gold copper AuCu 260.512512 ALLOY AUCU3 Gold 3-copper AuCu3 387.604492 ALLOY AUSN Gold tin AuSn 315.676514 ALLOY AUSN2 Gold 2-tin AuSn2 434.386505 ALLOY AUSN4 Gold 4-tin AuSn4 671.806519 ALLOY BA2SN 2-Barium tin Ba2Sn 393.369995 ALLOY BI2U 2-Bismuth uranium Bi2U 655.987671 ALLOY BI4U3 4-Bismuth 3-uranium Bi4U3 1550.002319 ALLOY BIU Bismuth uranium BiU 447.007294 ALLOY CA2PB 2-Calcium lead Ca2Pb 287.355988 ALLOY CA2SI 2-Calcium silicon Ca2Si 108.241501 ALLOY CA2SN 2-Calcium tin Ca2Sn 198.865997 ALLOY CA3SB2 3-Calcium 2-antimony Ca3Sb2 363.734009 ALLOY CAMG2 Calcium 2-magnesium CaMg2 88.688004 ALLOY CAPB Calcium lead CaPb 247.278 ALLOY CASI Calcium silicon CaSi 68.163498 ALLOY CASI2 Calcium 2-silicon CaSi2 96.249001 ALLOY CASN Calcium tin CaSn 158.787994 ALLOY CAZN Calcium zinc CaZn 105.468002 ALLOY CAZN2 Calcium 2-zinc CaZn2 170.858002 ALLOY CD11U 11-Cadmium uranium Cd11U 1474.536865 ALLOY CD3AS2 3-Cadmium 2-arsenic As2Cd3 487.073212 -

Barium Oxalate Single Crystals Grown in Silica Gel
Asian Journal of Chemistry Vol. 20, No. 8 (2008), 5971-5976 Characterization, Microhardness and Dielectric Studies of Pure and Doped (Copper, Cadmium) Barium Oxalate Single Crystals Grown in Silica Gel M. ABDUL SALIM* and R. RAVEENDRAN† Department of Physics, TKM College of Arts and Science, Kollam-691 005, India E-mail: [email protected] Single crystals of pure barium oxalate, copper doped barium oxalate and cadmium doped barium oxalate crystals are grown by gel method. The double diffusion (U-tube) method is adopted for growing crystals. The crystals are of few mm in size. 5-7 d taken for gel set and 45-60 d taken for complete growth of the crystals. The X-ray diffraction patterns of the samples revealed their crystalline nature. Thermo gravi- metric analysis data supported the release of lattice H2O, CO, and CO2. The FTIR spectrum of the crystals showed the presence of water molecules and carboxylic acid. The characteristics are slightly different due to the doping with impurities. The microhardness and dielectric studies of grown crystals were also carried out. Key Words: Doped barium oxalate crystals, Microhard- ness, Dielectric property. INTRODUCTION The varieties of scientifically and technologically important materials have increased tremendously in recent years. The oxalate crystals have more important roles in the urinary crystal fields. The small variations on characteristics and properties due to doping is clear in the analysis results. In present paper, growth, characterization and properties of pure barium oxalate and two doped barium oxalate crystals are tried. Growth by hydro- silica gel1 is the most suitable method for getting perfect crystals with minimum impurities and imperfections.