Comparing SNP Panels and Statistical Methods for Estimating Genomic Breed Composition of Individual Animals in Ten Cattle Breeds
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Improving Beef Cattle Performance on Tall Fescue
University of Tennessee, Knoxville TRACE: Tennessee Research and Creative Exchange Doctoral Dissertations Graduate School 5-2012 Improving Beef Cattle Performance on Tall Fescue Brian Thomas Campbell [email protected] Follow this and additional works at: https://trace.tennessee.edu/utk_graddiss Part of the Animal Sciences Commons Recommended Citation Campbell, Brian Thomas, "Improving Beef Cattle Performance on Tall Fescue. " PhD diss., University of Tennessee, 2012. https://trace.tennessee.edu/utk_graddiss/1277 This Dissertation is brought to you for free and open access by the Graduate School at TRACE: Tennessee Research and Creative Exchange. It has been accepted for inclusion in Doctoral Dissertations by an authorized administrator of TRACE: Tennessee Research and Creative Exchange. For more information, please contact [email protected]. To the Graduate Council: I am submitting herewith a dissertation written by Brian Thomas Campbell entitled "Improving Beef Cattle Performance on Tall Fescue." I have examined the final electronic copy of this dissertation for form and content and recommend that it be accepted in partial fulfillment of the requirements for the degree of Doctor of Philosophy, with a major in Animal Science. John C. Waller, Major Professor We have read this dissertation and recommend its acceptance: F. David Kirkpatrick, Gina M. Pighetti, Gary E. Bates, Cheryl J. Kojima Accepted for the Council: Carolyn R. Hodges Vice Provost and Dean of the Graduate School (Original signatures are on file with official studentecor r ds.) Improving Beef Cattle Performance on Tall Fescue Dissertation Presented for the Doctor of Philosophy Degree The University of Tennessee, Knoxville Brian Thomas Campbell May 2012 I Abstract The overall goal of the studies described in this dissertation was to improve beef production of cows grazing endophyte infected tall fescue either through management practices or through identifying markers for genetic selection. -

June 2019 Newsletter
Relationships Are a “I chose Santa Gertrudis cattle because I thought that’s what time, then sold at a local livestock sale in Unionville, Tennessee Fundamental Part cowboys ought to have,” Smith adds. “It’s more about the cattle “We’ve tried several ways to market the calves,” Warren says. culture for me but also that they are good cattle and take care of “We’ve put them in a cooperative, sent to a feedlot - it always de- themselves.” pends on the market. Typically, we sell a short time after wean- Ravenswood Farm’s cattle have always been Santa ing.” of the STAR 5 Gertrudis-influenced. They started with purebred Santa Gertrudis Warren adds that in the commercial beef cattle business, they but have moved to the crossbred STAR 5 cattle. Warren says the are trying to raise a calf that pushes the scale down as fast as it cattle from Corporron are gentle, raise a good calf and are good can, trying to get to a number as quickly as possible with the Stephen Smith (l) and Marty Warren (c), with Ravenswood Farm are Female Business milkers that haven’t had udder problems. least amount of expense. pictured with Jim Corporron at the San Antonio All Breed Bull and “We appreciate the Hereford-influence in the Santa Gertrudis Besides having growthy calves, Smith wants to have cattle Commercial Female Sale. Ravenswood Farm are repeat Star 5 fe- By Kelsey Pope, Freelance Writer, cattle for being gentle and raising good calves,” Warren says. that look good, too. males customers for the Corporron Family. -

Multiple Choice Choose the Answer That Best Completes Each Statement Or Question
Name Date Hour 5 The Beef Cattle Industry Multiple Choice Choose the answer that best completes each statement or question. _______ 1. Early settlers primarily used cattle as ____ . A. work animals B. a source of meat C. a source of milk D. symbols of wealth _______ 2. The modern cattle industry is concentrated in the ____ . A. South and Midwest B. South and Southwest C. North and Northwest D. North and Midwest _______ 3. Cattle drives were necessary in the past because of the lack of ____ . A. retail stores B. refrigeration C. slaughterhouses D. year-round grazing _______ 4. Which beef cattle breed is known for its solid black color and excellent meat quality? A. Angus B. Hereford C. Shorthorn D. Chianina _______ 5. Which beef cattle breed is from northern England and was often called a Durham after the county in which it originated? A. Angus B. Hereford C. Shorthorn D. Chianina Introduction to Agriscience | Unit 5 Test CIMC 1 _______ 6. Which beef cattle breed originated in England and was originally much larger, weighing more than 3,000 pounds, than it is today? A. Angus B. Hereford C. Shorthorn D. Chianina _______ 7. Which beef cattle breed is red with a white face and may also have white on the neck, underline, legs, and tail switch? A. Angus B. Hereford C. Shorthorn D. Chianina _______ 8. Which beef cattle breed is one of the oldest breeds in the world and originated in Italy? A. Angus B. Hereford C. Shorthorn D. Chianina _______ 9. Which beef cattle breed originated in central France and was developed as a dual-purpose breed and is typically white or off-white in color? A. -
Proc1-Beginning Chapters.Pmd
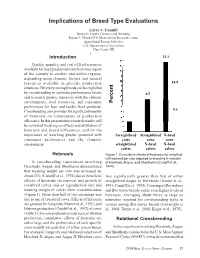
Implications of Breed Type Evaluations Larry V. Cundiff Research Leader, Genetics and Breeding Roman L. Hruska U.S. Meat Animal Research Center Agricultural Research Service U.S. Department of Agriculture Clay Center, NE ()(,) Introduction 23.3 Quality, quantity, and cost of feed resources available for beef production vary from one region of the country to another and within regions, depending upon climatic factors and natural 23.3 resources available in specific production 14.8 situations. Diversity among breeds can be exploited by crossbreeding to optimize performance levels 8.5 and to match genetic resources with the climatic environment, feed resources, and consumer Percent preferences for lean and tender beef products. 8.5 Crossbreeding also provides for significant benefits 14.8 of heterosis on components of production efficiency. In this presentation, research results will be reviewed focusing on effects and utilization of 8.5 8.5 heterosis and breed differences, and on the importance of matching genetic potential with Straightbred Straightbred X-bred consumer preferences and the climatic cows cows cows environment. straightbred X-bred X-bred calves calves calves Heterosis Figure 1. Cumulative effects of heterosis for weight of calf weaned per cow exposed to breeding in crosses A crossbreeding experiment involving of Hereford, Angus, and Shorthorns (Cundiff et al., Herefords, Angus, and Shorthorns demonstrated 1974). that weaning weight per cow was increased by about 23% (Cundiff et al., 1974) due to beneficial was significantly greater than that of either effects of heterosis on survival and growth of straightbred Angus or Herefords (Nunez et al., crossbred calves and on reproduction rate and 1991; Cundiff et al., 1992). -

Purebred Livestock Registry Associations
Purebred livestock registry associations W. Dennis Lamm1 COLORADO STATE UNIVERSITY EXTENSION SERVICE no. 1.217 Beef Devon. Devon Cattle Assn., Inc., P.O. Box 628, Uvalde, TX 78801. Mrs. Cammille Hoyt, Sec. Phone: American. American Breed Assn., Inc., 306 512-278-2201. South Ave. A, Portales, NM 88130. Mrs. Jewell Dexter. American Dexter Cattle Assn., P.O. Jones, Sec. Phone: 505-356-8019. Box 56, Decorah, IA 52l01. Mrs. Daisy Moore, Amerifax. Amerifax Cattle Assn., Box 149, Exec. Sec. Phone: 319-736-5772, Hastings, NE 68901. John Quirk, Pres. Phone Friesian. Beef Friesian Society, 213 Livestock 402-463-5289. Exchange Bldg., Denver, CO 80216. Maurice W. Angus. American Angus Assn., 3201 Freder- Boney, Adm. Dir. Phone: 303-587-2252. ick Blvd., St. Joseph, MO 64501. Richard Spader, Galloway. American Galloway Breeders Assn., Exec. Vice. Pres. Phone: 816-233-3101. 302 Livestock Exchange Bldg., Denver, CO 80216. Ankina. Ankina Breeders, Inc., 5803 Oaks Rd,. Cecil Harmon, Pres. Phone: 303-534-0853. Clayton, OH 45315. James K. Davis, Ph.D., Pres. Galloway. Galloway Cattle Society of Amer- Phone: 513-837-4128. ica, RFD 1, Springville, IA 52336. Phone: 319- Barzona. Barzona Breeders Assn. of America, 854-7062. P.O. Box 631, Prescott, AZ 86320. Karen Halford, Gelbvieh. American Gelbvieh Assn., 5001 Na- Sec. Phone: 602-445-2290. tional Western Dr., Denver, CO 80218. Daryl W. Beefalo. American Beefalo Breeders, 1661 E. Loeppke, Exec. Dir. Phone: 303-296-9257. Brown Rd., Mayville 22, MI 48744. Phone: 517-843- Hays Convertor. Canadian Hays Convertor 6811. Assn., 6707 Elbow Dr. SW, Suite 509, Calgary, Beefmaster. -

II. Outbreeding and Hybrid Vigour
II. Outbreeding and Hybrid vigour II. Outbreeding and Hybrid vigour • Outbreeding is the opposite of inbreeding, it is the mating of animals less closely related than the average relationship within the breed. • If two individuals do not have any ancestors in common for five or six generations back in their respective pedigrees, they are usually thought not being any more related than the average of the population Consequences of outbreeding 1. Outbreeding increases the number of pairs of heterozygous genes in the individual 2. Outbreeding tends to decrease breeding purity 3. Outbreeding tends to cover up detrimental recessive genes. 4. Phenotypically, outbreeding usually improves traits related to physical fitness (hybrid vigour). Hybrid vigour or heterosis • Heterosis, or hybrid vigour, is the name given to the increased vigour of the offspring over that of the parents when unrelated individuals are mated. • In 1914 Professor Shull proposed for the first time the word “heterosis ” • The best known example for hybrid vigour in animals is the mule (male ass and mare) • The reciprocal cross, called the hinney or jennet Types of heterosis • There are three main types of heterosis (Individual, Maternal, Paternal) 1) Individual (direct) heterosis • It is the improvement in the performance of crossbred individual above the average of its parents. • It is affected by Individual’s gene that directly affects on its performance. • All traits have what is called a direct or individual component of heterosis • Examples (weaning weight, yearling weight and carcass traits) 2- Maternal Heterosis • It is the improvement in the performance of the crossbred mother over the average of purebred mothers Example: I. -

Animal Genetic Resources Information Bulletin
The designations employed and the presentation of material in this publication do not imply the expression of any opinion whatsoever on the part of the Food and Agriculture Organization of the United Nations concerning the legal status of any country, territory, city or area or of its authorities, or concerning the delimitation of its frontiers or boundaries. Les appellations employées dans cette publication et la présentation des données qui y figurent n’impliquent de la part de l’Organisation des Nations Unies pour l’alimentation et l’agriculture aucune prise de position quant au statut juridique des pays, territoires, villes ou zones, ou de leurs autorités, ni quant au tracé de leurs frontières ou limites. Las denominaciones empleadas en esta publicación y la forma en que aparecen presentados los datos que contiene no implican de parte de la Organización de las Naciones Unidas para la Agricultura y la Alimentación juicio alguno sobre la condición jurídica de países, territorios, ciudades o zonas, o de sus autoridades, ni respecto de la delimitación de sus fronteras o límites. All rights reserved. No part of this publication may be reproduced, stored in a retrieval system, or transmitted in any form or by any means, electronic, mechanical, photocopying or otherwise, without the prior permission of the copyright owner. Applications for such permission, with a statement of the purpose and the extent of the reproduction, should be addressed to the Director, Information Division, Food and Agriculture Organization of the United Nations, Viale delle Terme di Caracalla, 00100 Rome, Italy. Tous droits réservés. Aucune partie de cette publication ne peut être reproduite, mise en mémoire dans un système de recherche documentaire ni transmise sous quelque forme ou par quelque procédé que ce soit: électronique, mécanique, par photocopie ou autre, sans autorisation préalable du détenteur des droits d’auteur. -

Florida Agriculture Statistical Directory
Dear Friends of Agriculture, It is my pleasure to present the 2008 Florida Agriculture Statistical Directory. This report presents a wealth of information about Florida’s vast and varied agricultural production through data that details land use, crop yields, commodity prices, crop rankings and more. This yearly report is invaluable to anyone who is involved in this dynamic business or who wants to better understand its complexities. The tables, charts and statistics contained in this report do an exceptional job of measuring the inputs and outputs, and presenting Florida agriculture in the context of “hard numbers.” But there is more to our state’s agricultural industry: our hard-working farmers, whose dedication, hard work and perseverance have made Florida agriculture into the diverse and highly productive industry that is respected throughout the globe. As evidenced by the ever-growing popularity of the “Fresh from Florida” label, consumers worldwide appreciate and seek out the quality products that our farmers provide. Maintaining these standards of excellence seldom comes easily as each year presents new challenges for Florida’s 40,000 commercial farmers. But, whether confronted by hurricanes, freezes, pests, diseases or fierce international competition, our state’s producers continually show that they are up to the test. Enterprising spirit, love of the land, and pride in their products are all hallmarks of the well- earned reputation of Florida’s farmers. In addition to enjoying the quality products that our farmers produce, Florida’s agricultural production benefits our state’s residents in other important ways as well. Florida agriculture has an overall economic impact estimated at more than $100 billion annually, making it a sound pillar of the state’s economy. -

ACE Appendix
CBP and Trade Automated Interface Requirements Appendix: PGA August 13, 2021 Pub # 0875-0419 Contents Table of Changes .................................................................................................................................................... 4 PG01 – Agency Program Codes ........................................................................................................................... 18 PG01 – Government Agency Processing Codes ................................................................................................... 22 PG01 – Electronic Image Submitted Codes .......................................................................................................... 26 PG01 – Globally Unique Product Identification Code Qualifiers ........................................................................ 26 PG01 – Correction Indicators* ............................................................................................................................. 26 PG02 – Product Code Qualifiers ........................................................................................................................... 28 PG04 – Units of Measure ...................................................................................................................................... 30 PG05 – Scientific Species Code ........................................................................................................................... 31 PG05 – FWS Wildlife Description Codes ........................................................................................................... -

American Registered Breeds (Arb) Other Registered
AMERICAN REGISTERED BREEDS (ARB) 1. Open to heifers of Bos indicus type that have been registered or issued a certificate of recordation with a recognized breed association. The breeds that will be recognized are defined below: • Beefmaster Advancer - Animals of fifty percent (50%) or more Registered Beefmaster breeding and fifty percent (50%) or less of other Registered and DNA genotyped non-Beefmaster Beef cattle breeding. • Braford - Purebred, Heifers must be classified as Braford to be eligible to show. No F-1, multiple generation half blood, ¾ Hereford, ¾ Brahman, Single Bar or other percentage cattle will be accepted. • Brangus Premium Gold – Progeny of Registered Brangus or Red Brangus and any commercial animal. Must maintain a minimum of 50% registered Brangus/Red Brangus genetics. • Brangus Optimizer – Progeny resulting from mating Registered Brangus or Red Brangus and a registered animal from another beef breed recognized by the U.S. Beef Breeds Council, other than Angus or Red Angus. Must maintain a minimum of 50% registered Brangus genetics. • Brangus UltraBlack – Progeny of Registered Brangus and an Enrolled Angus • Brangus UltraRed- Progeny of Registered Red Brangus and an Enrolled Angus/Red Angus • Certified Beefmaster E6 - Certified by Beefmaster Breeders United to be at least 50% Beefmaster and can be as much as 100%. At least one of the parents must be registered as a purebred Beefmaster. • Golden Certified F1 – A female that is the progeny of two registered parents with one parent being registered Brahman resulting in a F1 cross (50% Brahman x 50% Bos Taurus). Must be issued a certificate of recordation form from ABBA which includes owner’s name, ownership date. -

Updated ILRIC Standards Version 17 100309.Indd
and Quality Assurance Certification Process 1 2 PARTICIPATING BREEDS PARTICIPATING BREEDS TM AngusAUSTRALIA Belmont Red Santa Gertrudis Braford Lincoln Red Maine Anjou Australian Nguni Breeders Charolais 13 CONTENTS Introduction 3 1 Livestock Category Standards 4 2 Livestock Quality Assurance Inspection Standards 6 Structural Soundness Quality Assurance Specifi cation 7 Export Breeding Certifi cate Examples 8 Quality Assurance Certifi cation Process 10 3 Semen Standards 12 4 Embryo Standards 13 5 Individual Breed Specifi cations 14 Glossary of Terms 45 Contacts 49 2 4 CONTENTS INTRODUCTION The Australian Cattle Genetic Export Standards and Quality Assurance Certifi cation Process of the standards that are detailed in this document are produced by the Australian Cattle Genetics Export Agency (ACGEA) a wholly owned subsidiary of the International Livestock Resources and Information Centre (ILRIC) on behalf of the European, British and Tropical breeds as detailed in the document. The Standards have been endorsed by relevant industry peak bodies including the Australian Registered Cattle Breeders Association (ARCBA), Meat and Livestock Australia (MLA), the Australian Livestock Exporters Council (ALEC) and the Cattle Council of Australia (CCA). ILRIC also has as its members, 26 Australian Breed Associations. All breeding animals exported under The Australian Cattle Genetics Export Standard and Quality Assurance Certifi cation Process will be individually inspected and certifi ed by accredited ILRIC Inspectors. The certifi cation process is comprehensive and thorough. Animal identifi cation and pedigree data will be individually verifi ed and certifi ed as correct and in all categories there will be individual animal inspections in accordance with the compliance standard for structural soundness within the Australian structural soundness quality assurance specifi cations. -

Cattle Producer's Handbook
Western Beef Resource Committee Fourth Edition Cattle Producer’s Handbook Genetics Section 845 Breed Association Contact List J. Benton Glaze, Jr., University of Idaho Breed associations provide beef cattle producers a retain genetically superior animals for use in future variety of benefits and services. Breed associations work generations. To accomplish this task, producers must in the areas of breed promotion, marketing, member take advantage of available tools and resources, such education, performance recording, and performance as expected progeny differences (EPD). EPDs are an evaluation. While all services are important, one that evaluation of an animal’s genetic worth (value as a receives much attention is performance recording and parent). EPDs are reported in sire summaries, which are the evaluation of animals. published by several breed associations. To remain competitive in the beef cattle industry, Following is a list of breed associations and their producers must continually strive to identify and contact information. AMERIFAX BEEFALO Amerifax Cattle Association American Beefalo Association 400 N. Minnesota Ave. P.O. Box 295 P.O. Box 149 Benton City, WA 99320 Hastings, NE 68901 9824 E. YZ Ave. (402) 463-5289 Vicksburg, MI 49097 (800) 233-3256 ANGUS web: americanbeefalo.org American Angus Association 3201 Frederick Ave. BEEFMASTER St. Joseph, MO 64506 Beefmaster Breeders United (816) 383-5100 6800 Park Ten Blvd., Ste. 290W (816) 233-9703 fax San Antonio, TX 78213 web: www.angus.org (210) 732-3132 (210) 732-7711 fax BARZONA web: www.beefmasters.org Barzona Breeders Association of America 604 Cedar St. BLONDE D’AQUITAINE Adair, IA 50002 American Blonde D’Aquitaine Association (641) 745-9170 57 Friar Tuckway (641) 343-0927 fax Fyffe, AL 35971 web: www.barzona.com (256) 996-3142 web: www.blondecattle.org 845-1 BRAFORD GELBVIEH United Braford Breeders American Gelbvieh Association 638A N.