Binary and Ternary Transition-Metal Phosphides As Hydrodenitrogenation Catalysts
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

TR-499: Indium Phosphide (CASRN 22398-80-7) in F344/N Rats And
NTP TECHNICAL REPORT ON THE TOXICOLOGY AND CARCINOGENESIS STUDIES OF INDIUM PHOSPHIDE (CAS NO. 22398-80-7) IN F344/N RATS AND B6C3F1 MICE (INHALATION STUDIES) NATIONAL TOXICOLOGY PROGRAM P.O. Box 12233 Research Triangle Park, NC 27709 July 2001 NTP TR 499 NIH Publication No. 01-4433 U.S. DEPARTMENT OF HEALTH AND HUMAN SERVICES Public Health Service National Institutes of Health FOREWORD The National Toxicology Program (NTP) is made up of four charter agencies of the U.S. Department of Health and Human Services (DHHS): the National Cancer Institute (NCI), National Institutes of Health; the National Institute of Environmental Health Sciences (NIEHS), National Institutes of Health; the National Center for Toxicological Research (NCTR), Food and Drug Administration; and the National Institute for Occupational Safety and Health (NIOSH), Centers for Disease Control and Prevention. In July 1981, the Carcinogenesis Bioassay Testing Program, NCI, was transferred to the NIEHS. The NTP coordinates the relevant programs, staff, and resources from these Public Health Service agencies relating to basic and applied research and to biological assay development and validation. The NTP develops, evaluates, and disseminates scientific information about potentially toxic and hazardous chemicals. This knowledge is used for protecting the health of the American people and for the primary prevention of disease. The studies described in this Technical Report were performed under the direction of the NIEHS and were conducted in compliance with NTP laboratory health and safety requirements and must meet or exceed all applicable federal, state, and local health and safety regulations. Animal care and use were in accordance with the Public Health Service Policy on Humane Care and Use of Animals. -

Transition Metal Phosphides for the Catalytic Hydrodeoxygenation of Waste Oils Into Green Diesel
catalysts Review Transition Metal Phosphides for the Catalytic Hydrodeoxygenation of Waste Oils into Green Diesel M. Consuelo Alvarez-Galvan * , Jose M. Campos-Martin * and Jose L. G. Fierro * Energy and Sustainable Chemistry Group (EQS), Instituto de Catálisis y Petroleoquímica, CSIC, c/Marie Curie, 2 Cantoblanco, 28049 Madrid, Spain * Correspondence: [email protected] (M.C.A.-G.); [email protected] (J.M.C.-M.); jlgfi[email protected] (J.L.G.F.) Received: 28 February 2019; Accepted: 15 March 2019; Published: 22 March 2019 Abstract: Recently, catalysts based on transition metal phosphides (TMPs) have attracted increasing interest for their use in hydrodeoxygenation (HDO) processes destined to synthesize biofuels (green or renewable diesel) from waste vegetable oils and fats (known as hydrotreated vegetable oils (HVO)), or from bio-oils. This fossil-free diesel product is produced completely from renewable raw materials with exceptional quality. These efficient HDO catalysts present electronic properties similar to noble metals, are cost-efficient, and are more stable and resistant to the presence of water than other classical catalytic formulations used for hydrotreatment reactions based on transition metal sulfides, but they do not require the continuous supply of a sulfide source. TMPs develop a bifunctional character (metallic and acidic) and present tunable catalytic properties related to the metal type, phosphorous-metal ratio, support nature, texture properties, and so on. Here, the recent progress in TMP-based catalysts for HDO of waste oils is reviewed. First, the use of TMPs in catalysis is addressed; then, the general aspects of green diesel (from bio-oils or from waste vegetable oils and fats) production by HDO of nonedible oil compounds are presented; and, finally, we attempt to describe the main advances in the development of catalysts based on TMPs for HDO, with an emphasis on the influence of the nature of active phases and effects of phosphorous, promoters, and preparation methods on reactivity. -

Discovering Amorphous Indium Phosphide Nanostructures with High-Temperature Ab Initio Molecular Dynamics
Discovering Amorphous Indium Phosphide Nanostructures with High-Temperature ab Initio Molecular Dynamics The MIT Faculty has made this article openly available. Please share how this access benefits you. Your story matters. Citation Zhao, Qing, Lisi Xie, and Heather J. Kulik. “Discovering Amorphous Indium Phosphide Nanostructures with High-Temperature Ab Initio Molecular Dynamics.” The Journal of Physical Chemistry C 119.40 (2015): 23238–23249. As Published http://dx.doi.org/10.1021/acs.jpcc.5b07264 Publisher American Chemical Society (ACS) Version Author's final manuscript Citable link http://hdl.handle.net/1721.1/105155 Terms of Use Article is made available in accordance with the publisher's policy and may be subject to US copyright law. Please refer to the publisher's site for terms of use. Discovering Amorphous Indium Phosphide Nanostructures with High-Temperature Ab Initio Molecular Dynamics Qing Zhao1,2, Lisi Xie1, and Heather J. Kulik1,* 1Department of Chemical Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139 2Department of Mechanical Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139 ABSTRACT: We employ high-temperature ab initio molecular dynamics (AIMD) as a sampling approach to discover low-energy, semiconducting, indium phosphide nanostructures. Starting from under-coordinated models of InP (e.g. a single layer of InP(111)), rapid rearrangement into a stabilized, higher-coordinate but amorphous cluster is observed across the size range considered (In3P3 to In22P22). These clusters exhibit exponential decrease in energy per atom with system size as effective coordination increases, which we define through distance-cutoff coordination number assignment and partial charge analysis. The sampling approach is robust to initial configuration choice as consistent results are obtained when alternative crystal models or computationally efficient generation of structures from sequential addition and removal of atoms are employed. -

COMBINED LIST of Particularly Hazardous Substances
COMBINED LIST of Particularly Hazardous Substances revised 2/4/2021 IARC list 1 are Carcinogenic to humans list compiled by Hector Acuna, UCSB IARC list Group 2A Probably carcinogenic to humans IARC list Group 2B Possibly carcinogenic to humans If any of the chemicals listed below are used in your research then complete a Standard Operating Procedure (SOP) for the product as described in the Chemical Hygiene Plan. Prop 65 known to cause cancer or reproductive toxicity Material(s) not on the list does not preclude one from completing an SOP. Other extremely toxic chemicals KNOWN Carcinogens from National Toxicology Program (NTP) or other high hazards will require the development of an SOP. Red= added in 2020 or status change Reasonably Anticipated NTP EPA Haz list COMBINED LIST of Particularly Hazardous Substances CAS Source from where the material is listed. 6,9-Methano-2,4,3-benzodioxathiepin, 6,7,8,9,10,10- hexachloro-1,5,5a,6,9,9a-hexahydro-, 3-oxide Acutely Toxic Methanimidamide, N,N-dimethyl-N'-[2-methyl-4-[[(methylamino)carbonyl]oxy]phenyl]- Acutely Toxic 1-(2-Chloroethyl)-3-(4-methylcyclohexyl)-1-nitrosourea (Methyl-CCNU) Prop 65 KNOWN Carcinogens NTP 1-(2-Chloroethyl)-3-cyclohexyl-1-nitrosourea (CCNU) IARC list Group 2A Reasonably Anticipated NTP 1-(2-Chloroethyl)-3-cyclohexyl-1-nitrosourea (CCNU) (Lomustine) Prop 65 1-(o-Chlorophenyl)thiourea Acutely Toxic 1,1,1,2-Tetrachloroethane IARC list Group 2B 1,1,2,2-Tetrachloroethane Prop 65 IARC list Group 2B 1,1-Dichloro-2,2-bis(p -chloropheny)ethylene (DDE) Prop 65 1,1-Dichloroethane -
US Schedule for Internet V2
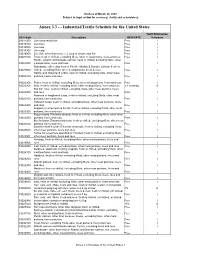
Draft as of March 23, 2007 Subject to legal review for accuracy, clarity and consistency. Annex 3.3 - - Industrial/Textile Schedule for the United States Tariff Elimination US 8 digit Description MFN RATE Schedule 03011000 Live ornamental fish Free I 03019100 Live trout Free I 03019200 Live eels Free I 03019300 Live carp Free I 03019900 Live fish, other than trout, eel, carp or ornamental fish Free I 03021100 Trout, fresh or chilled, excluding fillets, other meat portions, livers and roes Free I Pacific, Atlantic and Danube salmon, fresh or chilled, excluding fillets, other 03021200 meat portions, livers and roes Free I Salmonidae other than trout or Pacific, Atlantic & Danube salmon, fresh or 03021900 chilled, excluding fillets, other meat portions, livers & roes Free I Halibut and Greenland turbot, fresh or chilled, excluding fillets, other meat 03022100 portions, livers and roes Free I 03022200 Plaice, fresh or chilled, excluding fillets, other meat portions, livers and roes Free I 03022300 Sole, fresh or chilled, excluding fillets, other meat portions, livers and roes 1.1 cents/kg A Flat fish, nesi, fresh or chilled, excluding fillets, other meat portions, livers 03022900 and roes Free I Albacore or longfinned tunas, fresh or chilled, excluding fillets, other meat 03023100 portions, livers and roes Free I Yellowfin tunas, fresh or chilled, excluding fillets, other meat portions, livers 03023200 and roes Free I Skipjack or stripe-bellied bonito, fresh or chilled, excluding fillets, other meat 03023300 portions, livers and roes Free -

Study of Phosphorus Behaviour in Levitated Silicon-Iron Droplets
STUDY OF PHOSPHORUS BEHAVIOUR IN LEVITATED SILICON-IRON DROPLETS by Katherine Le A thesis submitted in conformity with the requirements for the degree of Master of Applied Science Department of Materials Science and Engineering University of Toronto © Copyright by Katherine Le 2016 ii Study of Phosphorus Behaviour in Levitated Si-Fe Droplets Katherine Le Master of Applied Science Department of Materials Science and Engineering University of Toronto 2016 Abstract While the treatment of relatively inexpensive ferrosilicon alloys is a potential refining route in order to generate solar grade silicon, phosphorus is one of the more difficult impurities to remove by conventional processing. In this project, electromagnetic levitation was used to investigate the dephosphorization of ferrosilicon alloy droplets exposed to H2-Ar gas mixtures under various experimental conditions including, refining time, temperature (1450°C-1720°C), H2-Ar gas concentrations and flow rate, iron alloying content, and initial phosphorus concentration. Reaction rates increased with higher refining times, temperatures, and H2 gas concentrations. With unknown parameters associated with the kinetics of gas phase reactions, the approach involved comparison of apparent activation energies derived for the chemical reaction and gas diffusion steps of the dephosphorization process. The phosphorus removal rate is thought to be controlled by the interfacial reaction step; further work is required to confirm this conclusion. iii Acknowledgements I would like to express my gratitude and respect to my supervisor, Prof. Alex McLean for the opportunity to work on this research. I am thankful for his guidance, wisdom and encouragement throughout the course of my studies. He is a truly inspiring person, and a great enabler of new learning opportunities. -

Red-Light Leds for Next-Generation Displays 6 July 2020
Red-light LEDs for next-generation displays 6 July 2020 indium gallium nitride (InGaN) semiconductors. Integrating two material systems is difficult. "Creating RGB displays requires the mass transfer of the separate blue, green and red LEDs together," says KAUST researcher Zhe Zhuang. An easier solution would be to create different-colored LEDs all on a single semiconductor chip. Optimizing the geometry, fabrication and electrical contacts is vital to maximizing the efficiency of the LED. Credit: Zhuag et al. Novel red LEDs are more temperature stable than those made using the conventional semiconductor of choice. In efforts to optimize the performance of light- emitting diodes (LEDs), King Abdullah University of Science and Technology researchers are looking at every aspect of the design, fabrication and operation of these devices. Now, they have succeeded in fabricating red LEDs, based on the The team developed an InGaN red LED structure where naturally blue-emitting semiconductor indium the output power is more stable than that of InGaP red gallium nitride, that are as stable as those based LEDs. Credit: Zhe Zhuang on indium gallium phosphide. LEDs are optical sources made from semiconductors that offer improvements on Since InGaP semiconductors are unable to emit conventional visible-light sources in terms of blue or green light, the only solution to making energy saving, smaller size and longer lifetimes. monolithic RGB micro-LEDs is to use InGaN. This LEDs can emit across the spectrum, from the material has the potential to shift its emission from ultraviolet to blue (B), green (G), red (R) and into blue to green, yellow and red by introducing more the infrared. -

Chemical Vapor Deposition of Heteroepitaxial Boron Phosphide Thin Films
University of Tennessee, Knoxville TRACE: Tennessee Research and Creative Exchange Doctoral Dissertations Graduate School 12-2013 Chemical Vapor Deposition of Heteroepitaxial Boron Phosphide Thin Films John Daniel Brasfield University of Tennessee - Knoxville, [email protected] Follow this and additional works at: https://trace.tennessee.edu/utk_graddiss Part of the Semiconductor and Optical Materials Commons Recommended Citation Brasfield, John Daniel, "Chemical aporV Deposition of Heteroepitaxial Boron Phosphide Thin Films. " PhD diss., University of Tennessee, 2013. https://trace.tennessee.edu/utk_graddiss/2558 This Dissertation is brought to you for free and open access by the Graduate School at TRACE: Tennessee Research and Creative Exchange. It has been accepted for inclusion in Doctoral Dissertations by an authorized administrator of TRACE: Tennessee Research and Creative Exchange. For more information, please contact [email protected]. To the Graduate Council: I am submitting herewith a dissertation written by John Daniel Brasfield entitled "Chemical Vapor Deposition of Heteroepitaxial Boron Phosphide Thin Films." I have examined the final electronic copy of this dissertation for form and content and recommend that it be accepted in partial fulfillment of the equirr ements for the degree of Doctor of Philosophy, with a major in Chemistry. Charles S. Feigerle, Major Professor We have read this dissertation and recommend its acceptance: Laurence Miller, Frank Vogt, Ziling Xue Accepted for the Council: Carolyn R. Hodges Vice Provost and Dean of the Graduate School (Original signatures are on file with official studentecor r ds.) Chemical Vapor Deposition of Heteroepitaxial Boron Phosphide Thin Films A Dissertation Presented for the Doctor of Philosophy Degree The University of Tennessee, Knoxville John Daniel Brasfield December 2013 Copyright © 2013 by John Daniel Brasfield All rights reserved. -

Reducing Bow of Ingap on Silicon Wafers
94 Technology focus: III-Vs on silicon Reducing bow of InGaP on silicon wafers Researchers use strain engineering without impacting dislocation density. esearchers based in Singapore and the USA have been working to control the wafer bow of Rindium gallium phosphide (InGaP) epitaxial lay- ers on 200mm silicon (Si) wafers [Bing Wang et al, Semicond. Sci. Technol., vol32, p125013, 2017]. “Based on these Wafer bow is caused by observations, we can stress arising mainly conclude that the from mismatches of threading dislocation coefficients of thermal expansion between densities of the InGaP InGaP, or other III-V wafers are not compound semiconduc- affected by the lattice tors, and silicon. The mismatch. Our Ge bow (more than 200µm buffers have similar in one recent report of gallium arsenide on threading dislocation 300mm silicon wafer) is density of 3x107/cm2. introduced when the The hetero-epitaxy of material cools after high- GaAs buffers and temperature epitaxial Figure 1. Epitaxial layer structure of three InGaP/Si deposition. Bowing InGaP films did not wafers. adversely affects wafer- increase the scale processing, partic- threading dislocation was by metal-organic chemical vapor deposition ularly for large-diameter density, which (MOCVD). The germanium on silicon template layer substrates. Wafer-scale was prepared separately in a two-step low/high-tem- indicates very good equipment typically perature process, using germane (GeH4) precursor. restricts the permitted epitaxy quality.” Plan-view transmission electron microscopy (PV-TEM) bow to less than 50µm. The team believes and etch pit density (EPD) analysis gave an estimate of 7 2 The team from the that the technique dislocation density of the order 3x10 /cm . -

Supporting Information For: Solution Phase Synthesis of Indium Gallium Phosphide Alloy Nanowires Nikolay Kornienko , Desiré D
Supporting Information for: Solution Phase Synthesis of Indium Gallium Phosphide Alloy Nanowires Nikolay Kornienko1, Desiré D. Whitmore1, Yi Yu1, Stephen R. Leone1,2,4, Peidong Yang*1,3,5,6 1Department of Chemistry, 2Department of Physics and 3Department of Materials Science Engineering, University of California, Berkeley 94720, United States 4Chemical Sciences Division and 5Materials Sciences Division, Lawrence Berkeley National Lab, Berkeley CA 94720, United States 6Kavli Energy Nanosciences Institute, Berkeley, California 94720, United States * Correspondence to: [email protected] Table of Contents: Figure S1. TEM In/Ga growth seeds ............................................................................................................................. 2 Figure S2. TEM of InP and GaP NWs ............................................................................................................................. 2 Figure S3. Composition and diameter control ......................................................................................................... 3 Figure S4. XRD of InxGa1‐xP NWs .................................................................................................................................. 4 Figure S5. Electron diffraction of InxGa1‐xP NWs ................................................................................................... 5 Figure S6. Raman spectra of InP and GaP .................................................................................................................. 6 -

Subject Index
SUBJECT INDEX Vol. 1: 1–698, Vol. 2: 699–1395, Vol. 3: 1397–2111, Vol. 4: 2113–2798, Vol. 5: 2799–3440. Page numbers suffixed by t and f refer to Tables and Figures respectively. Absorption spectra in aqueous solution, 752–770 of americium, 1364–1368 compounds of, 721–752 americium (III), 1364–1365, 1365f coordination complexes in solution, americium (IV), 1365 771–782 americium (V), 1366, 1367f history of, 699–700 americium (VI), 1366, 1367f isotope production, 702–703 americium (VII), 1367–1368, 1368f metallic state of, 717–721 of liquid plutonium, 963 in nature, 703–704 of neptunium, 763–766, 763f, 786–787 nuclear properties of, 700–702 neptunium (VII) ternary oxides, 729 separation and purification, 704–717 of plutonium plutonium hexafluoride, 1088, 1089f atomic properties of, 857–862 ions, 1113–1117, 1116t compounds of, 987–1108 plutonium (IV), 849 metal and intermetallic compounds of, polymerization, 1151, 1151f 862–987 tetrachloride, 1093–1094, 1094f natural occurrence of, 822–824 tribromide, 1099t, 1100 nuclear properties of, 815–822 trichloride, 1099, 1099t separation and purification of, 826–857 Accelerator mass spectrometry (AMS), of solution chemistry of, 1108–1203 neptunium, 790 Actinide elements Acetates atomic volumes of, 922–923, 923f of americium, 1322, 1323t extraction of of plutonium, 1177, 1180 DIDPA, 1276 Acetone, derived compounds, of americium, HDEHP, 1275 1322, 1323t, 1324 organophosphorus and Acids carbamoylphosphonate reagents, for Purex process, 711 1276–1278 for solvent extraction, 839 stripping of, 1280–1281 Actinide -

High Hazard Chemical Policy
Environmental Health & Safety Policy Manual Issue Date: 2/23/2011 Policy # EHS-200.09 High Hazard Chemical Policy 1.0 PURPOSE: To minimize hazardous exposures to high hazard chemicals which include select carcinogens, reproductive/developmental toxins, chemicals that have a high degree of toxicity. 2.0 SCOPE: The procedures provide guidance to all LSUHSC personnel who work with high hazard chemicals. 3.0 REPONSIBILITIES: 3.1 Environmental Health and Safety (EH&S) shall: • Provide technical assistance with the proper handling and safe disposal of high hazard chemicals. • Maintain a list of high hazard chemicals used at LSUHSC, see Appendix A. • Conduct exposure assessments and evaluate exposure control measures as necessary. Maintain employee exposure records. • Provide emergency response for chemical spills. 3.2 Principle Investigator (PI) /Supervisor shall: • Develop and implement a laboratory specific standard operation plan for high hazard chemical use per OSHA 29CFR 1910.1450 (e)(3)(i); Occupational Exposure to Hazardous Chemicals in Laboratories. • Notify EH&S of the addition of a high hazard chemical not previously used in the laboratory. • Ensure personnel are trained on specific chemical hazards present in the lab. • Maintain Material Safety Data Sheets (MSDS) for all chemicals, either on the computer hard drive or in hard copy. • Coordinate the provision of medical examinations, exposure monitoring and recordkeeping as required. 3.3 Employees: • Complete all necessary training before performing any work. • Observe all safety