Genomic Alterations Caused by HPV Integration in a Cohort of Chinese Endocervical Adenocarcinomas
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

C8cc08685k1.Pdf
Electronic Supplementary Material (ESI) for ChemComm. This journal is © The Royal Society of Chemistry 2018 Supporting Information Minimalist Linkers Suitable for Irreversible Inhibitors in Simultaneous Proteome Profiling, Live-Cell Imaging and Drug Screening Cuiping Guo,Yu Chang, Xin Wang, Chengqian Zhang, Piliang Hao*, Ke Ding and Zhengqiu Li* School of Pharmacy, Jinan University, Guangzhou, China 510632 *Corresponding author ([email protected]) 1. General Information All chemicals were purchased from commercial vendors and used without further purification, unless indicated otherwise. All reactions requiring anhydrous conditions were carried out under argon or nitrogen atmosphere using oven-dried glassware. AR-grade solvents were used for all reactions. Reaction progress was monitored by TLC on pre-coated silica plates (Merck 60 F254 nm, 0.25 µm) and spots were visualized by UV, iodine or other suitable stains. Flash column chromatography was carried out using silica gel (Qingdao Ocean). All NMR spectra (1H-NMR, 13C-NMR) were recorded on Bruker 300 MHz/400 MHz NMR spectrometers. Chemical shifts were reported in parts per million (ppm) referenced with respect to appropriate internal standards or residual solvent peaks (CDCl3 = 7.26 ppm, DMSO-d6 = 2.50 ppm). The following abbreviations were used in reporting spectra, br s (broad singlet), s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet), dd (doublet of doublets). Mass spectra were obtained on Agilent LC-ESI-MS system. All analytical HPLC were carried out on Agilent system. Water with 0.1% TFA and acetonitrile with 0.1% TFA were used as eluents and the flow rate was 0.5 mL/min. -

Identification of the Binding Partners for Hspb2 and Cryab Reveals
Brigham Young University BYU ScholarsArchive Theses and Dissertations 2013-12-12 Identification of the Binding arP tners for HspB2 and CryAB Reveals Myofibril and Mitochondrial Protein Interactions and Non- Redundant Roles for Small Heat Shock Proteins Kelsey Murphey Langston Brigham Young University - Provo Follow this and additional works at: https://scholarsarchive.byu.edu/etd Part of the Microbiology Commons BYU ScholarsArchive Citation Langston, Kelsey Murphey, "Identification of the Binding Partners for HspB2 and CryAB Reveals Myofibril and Mitochondrial Protein Interactions and Non-Redundant Roles for Small Heat Shock Proteins" (2013). Theses and Dissertations. 3822. https://scholarsarchive.byu.edu/etd/3822 This Thesis is brought to you for free and open access by BYU ScholarsArchive. It has been accepted for inclusion in Theses and Dissertations by an authorized administrator of BYU ScholarsArchive. For more information, please contact [email protected], [email protected]. Identification of the Binding Partners for HspB2 and CryAB Reveals Myofibril and Mitochondrial Protein Interactions and Non-Redundant Roles for Small Heat Shock Proteins Kelsey Langston A thesis submitted to the faculty of Brigham Young University in partial fulfillment of the requirements for the degree of Master of Science Julianne H. Grose, Chair William R. McCleary Brian Poole Department of Microbiology and Molecular Biology Brigham Young University December 2013 Copyright © 2013 Kelsey Langston All Rights Reserved ABSTRACT Identification of the Binding Partners for HspB2 and CryAB Reveals Myofibril and Mitochondrial Protein Interactors and Non-Redundant Roles for Small Heat Shock Proteins Kelsey Langston Department of Microbiology and Molecular Biology, BYU Master of Science Small Heat Shock Proteins (sHSP) are molecular chaperones that play protective roles in cell survival and have been shown to possess chaperone activity. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Divergence of Human and Mouse Brain Transcriptome Highlights Alzheimer Disease Pathways
Divergence of human and mouse brain transcriptome highlights Alzheimer disease pathways Jeremy A. Millera,b, Steve Horvathc,d, and Daniel H. Geschwindb,c,1 aInterdepartmental Program for Neuroscience, Departments of cHuman Genetics and dBiostatistics, and bProgram in Neurogenetics and Neurobehavioral Genetics, Department of Neurology and Semel Institute, University of California, Los Angeles, CA 90095-1769 Edited by Joseph S. Takahashi, Howard Hughes Medical Institute, University of Texas Southwestern Medical Center, Dallas, TX, and approved June 1, 2010 (received for review December 10, 2009) Because mouse models play a crucial role in biomedical research works by merging data from many microarray studies, and then related to the human nervous system, understanding the similari- applying weighted gene coexpression network analysis (WGCNA) ties and differences between mouse and human brain is of (4–7). WGCNA elucidates the higher-order relationships between fundamental importance. Studies comparing transcription in hu- genes based on their coexpression relationships, delineating modules man and mouse have come to varied conclusions, in part because of of biologically related genes and permitting a robust view of their relatively small sample sizes or underpowered methodologies. transcriptome organization (3, 5, 8, 9). Within groups of highly coexpressed genes (“modules”) that comprise the core functional To better characterize gene expression differences between mouse fi and human, we took a systems-biology approach by using weighted units of the transcriptional network, WGCNA also identi es gene coexpression network analysis on more than 1,000 micro- the most highly connected, or most central genes within each module, referred to as “hubs.” We find that both gene expression arrays from brain. -

Associated 16P11.2 Deletion in Drosophila Melanogaster
ARTICLE DOI: 10.1038/s41467-018-04882-6 OPEN Pervasive genetic interactions modulate neurodevelopmental defects of the autism- associated 16p11.2 deletion in Drosophila melanogaster Janani Iyer1, Mayanglambam Dhruba Singh1, Matthew Jensen1,2, Payal Patel 1, Lucilla Pizzo1, Emily Huber1, Haley Koerselman3, Alexis T. Weiner 1, Paola Lepanto4, Komal Vadodaria1, Alexis Kubina1, Qingyu Wang 1,2, Abigail Talbert1, Sneha Yennawar1, Jose Badano 4, J. Robert Manak3,5, Melissa M. Rolls1, Arjun Krishnan6,7 & 1234567890():,; Santhosh Girirajan 1,2,8 As opposed to syndromic CNVs caused by single genes, extensive phenotypic heterogeneity in variably-expressive CNVs complicates disease gene discovery and functional evaluation. Here, we propose a complex interaction model for pathogenicity of the autism-associated 16p11.2 deletion, where CNV genes interact with each other in conserved pathways to modulate expression of the phenotype. Using multiple quantitative methods in Drosophila RNAi lines, we identify a range of neurodevelopmental phenotypes for knockdown of indi- vidual 16p11.2 homologs in different tissues. We test 565 pairwise knockdowns in the developing eye, and identify 24 interactions between pairs of 16p11.2 homologs and 46 interactions between 16p11.2 homologs and neurodevelopmental genes that suppress or enhance cell proliferation phenotypes compared to one-hit knockdowns. These interac- tions within cell proliferation pathways are also enriched in a human brain-specific network, providing translational relevance in humans. Our study indicates a role for pervasive genetic interactions within CNVs towards cellular and developmental phenotypes. 1 Department of Biochemistry and Molecular Biology, The Pennsylvania State University, University Park, PA 16802, USA. 2 Bioinformatics and Genomics Program, The Huck Institutes of the Life Sciences, The Pennsylvania State University, University Park, PA 16802, USA. -

Genome-Wide Identification, Characterization and Expression
G C A T T A C G G C A T genes Article Genome-Wide Identification, Characterization and Expression Profiling of myosin Family Genes in Sebastes schlegelii Chaofan Jin 1, Mengya Wang 1,2, Weihao Song 1 , Xiangfu Kong 1, Fengyan Zhang 1, Quanqi Zhang 1,2,3 and Yan He 1,2,* 1 MOE Key Laboratory of Molecular Genetics and Breeding, College of Marine Life Sciences, Ocean University of China, Qingdao 266003, China; [email protected] (C.J.); [email protected] (M.W.); [email protected] (W.S.); [email protected] (X.K.); [email protected] (F.Z.); [email protected] (Q.Z.) 2 Laboratory of Tropical Marine Germplasm Resources and Breeding Engineering, Sanya Oceanographic Institution, Ocean University of China, Sanya 572000, China 3 Laboratory for Marine Fisheries Science and Food Production Processes, Qingdao National Laboratory for Marine Science and Technology, Qingdao 266003, China * Correspondence: [email protected]; Tel.: +86-0532-82031986 Abstract: Myosins are important eukaryotic motor proteins that bind actin and utilize the energy of ATP hydrolysis to perform a broad range of functions such as muscle contraction, cell migration, cytokinesis, and intracellular trafficking. However, the characterization and function of myosin is poorly studied in teleost fish. In this study, we identified 60 myosin family genes in a marine teleost, black rockfish (Sebastes schlegelii), and further characterized their expression patterns. myosin showed divergent expression patterns in adult tissues, indicating they are involved in different types and Citation: Jin, C.; Wang, M.; Song, W.; compositions of muscle fibers. Among 12 subfamilies, S. schlegelii myo2 subfamily was significantly Kong, X.; Zhang, F.; Zhang, Q.; He, Y. -

IDENTIFICATION and CHARACTERIZATION of ACTIN-REGULATORY PROTEINS in the HAIR CELL's CUTICULAR PLATE by LANA MARY POLLOCK Subm
IDENTIFICATION AND CHARACTERIZATION OF ACTIN-REGULATORY PROTEINS IN THE HAIR CELL’S CUTICULAR PLATE by LANA MARY POLLOCK Submitted in partial fulfilment of the requirements for the degree of Doctor of Philosophy Dissertation advisor: Brian M. McDermott Jr., Ph.D. Department of Genetics and Genome Sciences CASE WESTERN RESERVE UNIVERSITY January 2016 Case Western Reserve University School of Graduate Studies We, the thesis committee, hereby approve the thesis/dissertation of Lana Pollock, candidate for the degree of Doctor of Philosophy (PhD).* (signed)_________Zhenghe Wang, Ph.D._________________ (chair of committee) ___________Brian McDermott, Ph.D._______________ ___________ Hua Lou, Ph.D._____________________ ___________Stephen Maricich, Ph.D., M.D.___________ ___________Anthony Wynshaw-Boris, Ph.D., M.D._____ Date of defense_____September 8th, 2015_______________ *we also certify that written approval has been obtained for release of any proprietary material contained therein 2 This thesis is dedicated to Daniel Margevicius. Thank you for your unwavering love and support. Ačiū!! 3 Table of contents List of Tables ........................................................................................................ 7 List of Figures ....................................................................................................... 8 List of abbreviations ............................................................................................ 13 Abstract ............................................................................................................. -

Supp Table 6.Pdf
Supplementary Table 6. Processes associated to the 2037 SCL candidate target genes ID Symbol Entrez Gene Name Process NM_178114 AMIGO2 adhesion molecule with Ig-like domain 2 adhesion NM_033474 ARVCF armadillo repeat gene deletes in velocardiofacial syndrome adhesion NM_027060 BTBD9 BTB (POZ) domain containing 9 adhesion NM_001039149 CD226 CD226 molecule adhesion NM_010581 CD47 CD47 molecule adhesion NM_023370 CDH23 cadherin-like 23 adhesion NM_207298 CERCAM cerebral endothelial cell adhesion molecule adhesion NM_021719 CLDN15 claudin 15 adhesion NM_009902 CLDN3 claudin 3 adhesion NM_008779 CNTN3 contactin 3 (plasmacytoma associated) adhesion NM_015734 COL5A1 collagen, type V, alpha 1 adhesion NM_007803 CTTN cortactin adhesion NM_009142 CX3CL1 chemokine (C-X3-C motif) ligand 1 adhesion NM_031174 DSCAM Down syndrome cell adhesion molecule adhesion NM_145158 EMILIN2 elastin microfibril interfacer 2 adhesion NM_001081286 FAT1 FAT tumor suppressor homolog 1 (Drosophila) adhesion NM_001080814 FAT3 FAT tumor suppressor homolog 3 (Drosophila) adhesion NM_153795 FERMT3 fermitin family homolog 3 (Drosophila) adhesion NM_010494 ICAM2 intercellular adhesion molecule 2 adhesion NM_023892 ICAM4 (includes EG:3386) intercellular adhesion molecule 4 (Landsteiner-Wiener blood group)adhesion NM_001001979 MEGF10 multiple EGF-like-domains 10 adhesion NM_172522 MEGF11 multiple EGF-like-domains 11 adhesion NM_010739 MUC13 mucin 13, cell surface associated adhesion NM_013610 NINJ1 ninjurin 1 adhesion NM_016718 NINJ2 ninjurin 2 adhesion NM_172932 NLGN3 neuroligin -
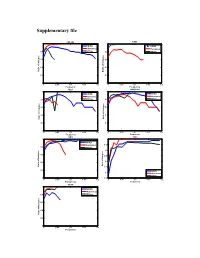
Supplementary File
Supplementary file COL1A2 FZD3 1 1 HPRD HPRD Humannet Humannet 0.8 PPIwu 0.8 PPIwu 0.6 0.6 0.4 0.4 Ratio of of indegree Ratio Ratio of Indegree 0.2 0.2 0 0 0 0.05 0.1 0.15 0.2 0 0.05 0.1 0.15 0.2 Frequency Frequency RBL1 SMARCA4 1 1 HPRD HPRD Humannet Humannet 0.8 PPIwu 0.8 PPIwu 0.6 0.6 0.4 0.4 Ratio of Indegree Ratio of Indegree Ratio 0.2 0.2 0 0 0 0.05 0.1 0.15 0.2 0 0.05 0.1 0.15 0.2 Frequency Frequency SRC TP53 1 1 HPRD Humannet 0.95 0.8 PPIwu 0.9 e 0.6 0.85 0.8 0.4 Ratio of Indegree of Ratio Ratio of Indegre 0.75 0.2 HPRD 0.7 Humannet PPIwu 0 0.65 0 0.05 0.1 0.15 0.2 0 0.05 0.1 0.15 0.2 Frequency Frequency VCAN 1 HPRD Humannet 0.8 PPIwu 0.6 0.4 Ratio of Indegree 0.2 0 0 0.05 0.1 0.15 0.2 Frequency Figure S1. Variation of indegree ratio with frequency cutoffs from 0 to 0.2 with a step 0.002 for driver genes common by three fitness cores in COAD. The indegree ratio is set to NULL if total degree of this genes is less than 5. BRAF CASR 1 1 0.8 0.8 0.6 0.6 0.4 0.4 Ratio of Indegree Ratio of Indegree of Ratio HPRD HPRD 0.2 0.2 Humannet Humannet PPIwu PPIwu 0 0 0 0.05 0.1 0.15 0.2 0 0.05 0.1 0.15 0.2 Frequency Frequency HDAC9 NRAS 1 1 0.8 0.8 0.6 0.6 0.4 0.4 HPRD HPRD Ratio of Indegree Ratio of Indegree 0.2 0.2 Humannet Humannet PPIwu PPIwu 0 0 0 0.05 0.1 0.15 0.2 0 0.05 0.1 0.15 0.2 Frequency Frequency NF1 CNTNAP2 1 1 0.9 0.8 0.8 0.6 0.7 0.4 HPRD HPRD Ratio of Indegree Ratio of Indegree 0.6 Humannet 0.2 Humannet PPIwu PPIwu 0.5 0 0 0.05 0.1 0.15 0.2 0 0.05 0.1 0.15 0.2 Frequency Frequency Figure S2. -

The Human Genome Project
TO KNOW OURSELVES ❖ THE U.S. DEPARTMENT OF ENERGY AND THE HUMAN GENOME PROJECT JULY 1996 TO KNOW OURSELVES ❖ THE U.S. DEPARTMENT OF ENERGY AND THE HUMAN GENOME PROJECT JULY 1996 Contents FOREWORD . 2 THE GENOME PROJECT—WHY THE DOE? . 4 A bold but logical step INTRODUCING THE HUMAN GENOME . 6 The recipe for life Some definitions . 6 A plan of action . 8 EXPLORING THE GENOMIC LANDSCAPE . 10 Mapping the terrain Two giant steps: Chromosomes 16 and 19 . 12 Getting down to details: Sequencing the genome . 16 Shotguns and transposons . 20 How good is good enough? . 26 Sidebar: Tools of the Trade . 17 Sidebar: The Mighty Mouse . 24 BEYOND BIOLOGY . 27 Instrumentation and informatics Smaller is better—And other developments . 27 Dealing with the data . 30 ETHICAL, LEGAL, AND SOCIAL IMPLICATIONS . 32 An essential dimension of genome research Foreword T THE END OF THE ROAD in Little has been rapid, and it is now generally agreed Cottonwood Canyon, near Salt that this international project will produce Lake City, Alta is a place of the complete sequence of the human genome near-mythic renown among by the year 2005. A skiers. In time it may well And what is more important, the value assume similar status among molecular of the project also appears beyond doubt. geneticists. In December 1984, a conference Genome research is revolutionizing biology there, co-sponsored by the U.S. Department and biotechnology, and providing a vital of Energy, pondered a single question: Does thrust to the increasingly broad scope of the modern DNA research offer a way of detect- biological sciences. -

Myosin IIIB Uses an Actin-Binding Motif in Its Espin-1 Cargo to Reach the Tips of Actin Protrusions
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector Current Biology 22, 320–325, February 21, 2012 ª2012 Elsevier Ltd All rights reserved DOI 10.1016/j.cub.2011.12.053 Report Myosin IIIB Uses an Actin-Binding Motif in Its Espin-1 Cargo to Reach the Tips of Actin Protrusions Raymond C. Merritt,1,2,5 Uri Manor,1,5 Felipe T. Salles,1,5 domain (THDII; [3]), failed to localize to filopodia tips (Fig- M’hamed Grati,1 Andrea C. Dose,3 William C. Unrath,4 ure 1A). As predicted by the inchworm hypothesis that Omar A. Quintero,4 Christopher M. Yengo,4 depends on the actin-binding activity of the THDII [1], and Bechara Kachar1,* MYO3A lacking the THDII (GFP-MYO3ADTHDII) also failed to 1Laboratory of Cell Structure and Dynamics, National Institute localize to filopodia tips (Figure 1A). However, when we fused on Deafness and Other Communication Disorders, a THDII from MYO3A onto the C terminus of MYO3B (cherry- National Institutes of Health, Bethesda, MD 20892, USA MYO3B:THDII), the hybrid protein localized to the tips of filo- 2Department of Biology, Gallaudet University, Washington, podia (Figure 1A). DC 20002, USA To confirm that the THDII interacts with actin in cells, we 3Department of Molecular and Cell Biology, University of transfected COS7 cells with GFP tagged MYO3A tail California, Berkeley, Berkeley, CA 94720, USA constructs containing the THDII, and we found that the GFP 4Department of Cellular and Molecular Physiology, Penn State fluorescence colocalized with actin (Figure 1B). -

1 SUPPLEMENTARY INFORMATION Supplementary Information Related
SUPPLEMENTARY INFORMATION Supplementary figure 1, related to figure 1: a. Gating strategy used to analyse FDG+/Lyl-1+ expression in MΦ progenitors from E9 and E9.5 YS from WT, Lyl-1WT/LacZ and Lyl-1LacZ/LacZ embryos (Fig. 1b) and in MΦ progenitors and mature MΦ progenitors from E10 WT and Lyl-1LacZ/LacZ YS, FL and brain (Fig. 3a, 4a, c, g). Shown is WT YS sample at E10. b. Gene set enrichment analysis (GSEA) of the whole transcriptome of WT E9 YS MΦ progenitors, compared to the EMP (left panel) and the MΦ (right panel) signatures defined by (Mass et al., 2016) (NES: normalised enrichment score; FDR: false discovery rate). c. Relative expression levels of factors thought to characterise either primitive (Lyl-1) or transient definitive progenitors (cMyb, Tlr2). A tendency to decreases (Lyl-1) or to increased (cMyb, Tlr2) at E10 is consistent with their wave of origin, albeit at non-significant levels due to the presence of progenitors from both waves in E10 YS (p-values were determined by unpaired, two-tailed t-Test). d. Top 1 IPA network representing the molecular relationships between DEG in E9 vs E10 WT MΦ progenitors indicated an overexpression of MHC-II at E9, while E10 progenitors over-expressed the TNFα signalling pathway. e. Flow cytometry analysis of MHC-II expression in MΦ progenitors from E9 and E10 WT YS. MHC-II expression is significantly reduced in E10 CD45+CD11b+c-Kit+ MΦ progenitors compared to E9, irrelevant of the genotype. f. Top 5 IPA network Illustrates the bias towards inflammatory signalling in E10 WT MΦ progenitors, compared to those present at E9.