Correction of a Splicing Mutation Affecting an Unverricht-Lundborg Disease Patient by Antisense Therapy
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Cystatins in Immune System
Journal of Cancer 2013, Vol. 4 45 Ivyspring International Publisher Journal of Cancer 2013; 4(1): 45-56. doi: 10.7150/jca.5044 Review Cystatins in Immune System Špela Magister1 and Janko Kos1,2 1. Jožef Stefan Institute, Department of Biotechnology, Ljubljana, Slovenia; 2. University of Ljubljana, Faculty of Pharmacy, Ljubljana, Slovenia. Corresponding author: Janko Kos, Ph. D., Department of Biotechnology, Jožef Stefan Institute, &Faculty of Pharmacy, University of Ljubljana, Slovenia; [email protected]; Phone+386 1 4769 604, Fax +3861 4258 031. © Ivyspring International Publisher. This is an open-access article distributed under the terms of the Creative Commons License (http://creativecommons.org/ licenses/by-nc-nd/3.0/). Reproduction is permitted for personal, noncommercial use, provided that the article is in whole, unmodified, and properly cited. Received: 2012.10.22; Accepted: 2012.12.01; Published: 2012.12.20 Abstract Cystatins comprise a large superfamily of related proteins with diverse biological activities. They were initially characterised as inhibitors of lysosomal cysteine proteases, however, in recent years some alternative functions for cystatins have been proposed. Cystatins pos- sessing inhibitory function are members of three families, family I (stefins), family II (cystatins) and family III (kininogens). Stefin A is often linked to neoplastic changes in epithelium while another family I cystatin, stefin B is supposed to have a specific role in neuredegenerative diseases. Cystatin C, a typical type II cystatin, is expressed in a variety of human tissues and cells. On the other hand, expression of other type II cystatins is more specific. Cystatin F is an endo/lysosome targeted protease inhibitor, selectively expressed in immune cells, suggesting its role in processes related to immune response. -

Anti-Cystatin B / Stefin B Antibody (ARG56897)
Product datasheet [email protected] ARG56897 Package: 100 μl anti-Cystatin B / Stefin B antibody Store at: -20°C Summary Product Description Rabbit Polyclonal antibody recognizes Cystatin B / Stefin B Tested Reactivity Hu Tested Application ICC/IF, IHC-P, WB Host Rabbit Clonality Polyclonal Isotype IgG Target Name Cystatin B / Stefin B Antigen Species Human Immunogen Recombinant fusion protein corresponding to aa. 1-98 of Human Cystatin B / Stefin B (NP_000091.1). Conjugation Un-conjugated Alternate Names Liver thiol proteinase inhibitor; EPM1; CPI-B; EPM1A; Cystatin-B; Stefin-B; PME; CST6; ULD; STFB Application Instructions Application table Application Dilution ICC/IF 1:50 - 1:200 IHC-P 1:50 - 1:200 WB 1:500 - 1:2000 Application Note * The dilutions indicate recommended starting dilutions and the optimal dilutions or concentrations should be determined by the scientist. Positive Control MCF7 and DU145 Calculated Mw 11 kDa Observed Size 14 kDa Properties Form Liquid Purification Affinity purified. Buffer PBS (pH 7.3), 0.02% Sodium azide and 50% Glycerol. Preservative 0.02% Sodium azide Stabilizer 50% Glycerol Storage instruction For continuous use, store undiluted antibody at 2-8°C for up to a week. For long-term storage, aliquot and store at -20°C. Storage in frost free freezers is not recommended. Avoid repeated freeze/thaw cycles. Suggest spin the vial prior to opening. The antibody solution should be gently mixed before use. www.arigobio.com 1/3 Note For laboratory research only, not for drug, diagnostic or other use. Bioinformation Gene Symbol CSTB Gene Full Name cystatin B (stefin B) Background The cystatin superfamily encompasses proteins that contain multiple cystatin-like sequences. -

Genomic Analysis of Bone Marrow Failure and Myelodysplastic Syndromes Reveals Phenotypic and Diagnostic Complexity
Bone Marrow Failure SUPPLEMENTARY APPENDIX Genomic analysis of bone marrow failure and myelodysplastic syndromes reveals phenotypic and diagnostic complexity Michael Y. Zhang,1 Siobán B. Keel,2 Tom Walsh,3 Ming K. Lee,3 Suleyman Gulsuner,3 Amanda C. Watts,3 Colin C. Pritchard,4 Stephen J. Salipante,4 Michael R. Jeng,5 Inga Hofmann,6 David A. Williams,6,7 Mark D. Fleming,8 Janis L. Abkowitz,2 Mary-Claire King,3 and Akiko Shimamura1,9,10 M.Y.Z. and S.B.K. contributed equally to this work. 1Clinical Research Division, Fred Hutchinson Cancer Research Center, Seattle, WA; 2Department of Medicine, Division of Hematology, University of Washington, Seattle, WA; 3Department of Medicine and Department of Genome Sciences, University of Washington, Seattle, WA; 4Department of Laboratory Medicine, University of Washington, Seattle, WA; 5Department of Pediatrics, Stanford Uni- versity School of Medicine, Stanford, CA; 6Division of Hematology/Oncology, Boston Children’s Hospital, Dana Farber Cancer Insti- tute, and Harvard Medical School, Boston, MA; 7Harvard Stem Cell Institute, Boston, MA; 8Department of Pathology, Boston Children’s Hospital, MA; 9Department of Pediatric Hematology/Oncology, Seattle Children’s Hospital, WA; 10Department of Pedi- atrics, University of Washington, Seattle, WA, USA ©2014 Ferrata Storti Foundation. This is an open-access paper. doi:10.3324/haematol.2014.113456 Manuscript received on July 22, 2014. Manuscript accepted on September 15, 2014. Correspondence: [email protected] Supplementary Methods Genomics. Libraries were prepared in 96-well format with a Bravo liquid handling robot (Agilent Technologies). One to two micrograms of genomic DNA were sheared to a peak size of 150 bp using a Covaris E series instrument. -
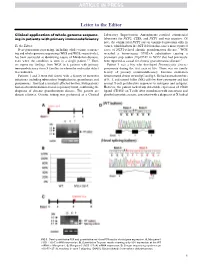
Clinical Application of Whole-Genome Sequencing in Patients with Primary
Letter to the Editor Clinical application of whole-genome sequenc- Laboratory Improvement Amendments–certified commercial ing in patients with primary immunodeficiency laboratory for NCF2, CYBA, and NCF1 and was negative. Of note, the commercial NCF1 screen examined mutations only in To the Editor: exon 2, which harbors the 2GT deletion that causes most reported Next-generation sequencing, including whole-exome sequenc- cases of NCF1-related chronic granulomatous disease.4 WGS ing and whole-genome sequencing (WES and WGS, respectively), revealed a homozygous 579G>A substitution causing a has been successful at identifying causes of Mendelian diseases, premature stop codon (Trp193X) in NCF1 that had previously even when the condition is seen in a single patient.1-3 Here, been reported as causal for chronic granulomatous disease.5 we report our findings from WGS in 6 patients with primary Patient 3 was a boy who developed Pneumocystis jiroveci immunodeficiency from 5 families in whom the molecular defect pneumonia during the first year of life. There was no family was unknown. history of primary immunodeficiency. Immune evaluation Patients 1 and 2 were full sisters with a history of recurrent demonstrated absent serum IgG and IgA. He had normal numbers infections, including tuberculous lymphadenitis, granulomas, and of B, T, and natural killer (NK) cells by flow cytometry and had pneumonias. They had a similarly affected brother. Both patients normal T-cell proliferative responses to mitogens and antigens. had an absent rhodamine-based respiratory burst, confirming the However, the patient lacked any detectable expression of CD40 diagnosis of chronic granulomatous disease. The parents are ligand (CD154) on T cells after stimulation with ionomycin and distant relatives. -
![Stefin B (CSTB) Mouse Monoclonal Antibody [Clone ID: OTI1E8] Product Data](https://docslib.b-cdn.net/cover/1226/stefin-b-cstb-mouse-monoclonal-antibody-clone-id-oti1e8-product-data-161226.webp)
Stefin B (CSTB) Mouse Monoclonal Antibody [Clone ID: OTI1E8] Product Data
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for TA813046 Stefin B (CSTB) Mouse Monoclonal Antibody [Clone ID: OTI1E8] Product data: Product Type: Primary Antibodies Clone Name: OTI1E8 Applications: IHC, WB Recommended Dilution: WB 1:500, IHC 1:500 Reactivity: Human Host: Mouse Isotype: IgG1 Clonality: Monoclonal Immunogen: Human recombinant protein fragment corresponding to amino acids 1-98 of human CSTB (NP_000091) produced in E.coli. Formulation: PBS (PH 7.3) containing 1% BSA, 50% glycerol and 0.02% sodium azide. Concentration: 1 mg/ml Purification: Purified from mouse ascites fluids or tissue culture supernatant by affinity chromatography (protein A/G) Conjugation: Unconjugated Storage: Store at -20°C as received. Stability: Stable for 12 months from date of receipt. Predicted Protein Size: 11 kDa Gene Name: cystatin B Database Link: NP_000091 Entrez Gene 1476 Human P04080 This product is to be used for laboratory only. Not for diagnostic or therapeutic use. View online » ©2021 OriGene Technologies, Inc., 9620 Medical Center Drive, Ste 200, Rockville, MD 20850, US 1 / 2 Stefin B (CSTB) Mouse Monoclonal Antibody [Clone ID: OTI1E8] – TA813046 Background: The cystatin superfamily encompasses proteins that contain multiple cystatin-like sequences. Some of the members are active cysteine protease inhibitors, while others have lost or perhaps never acquired this inhibitory activity. There are three inhibitory families in the superfamily, including the type 1 cystatins (stefins), type 2 cystatins and kininogens. This gene encodes a stefin that functions as an intracellular thiol protease inhibitor. -

1 Supporting Information for a Microrna Network Regulates
Supporting Information for A microRNA Network Regulates Expression and Biosynthesis of CFTR and CFTR-ΔF508 Shyam Ramachandrana,b, Philip H. Karpc, Peng Jiangc, Lynda S. Ostedgaardc, Amy E. Walza, John T. Fishere, Shaf Keshavjeeh, Kim A. Lennoxi, Ashley M. Jacobii, Scott D. Rosei, Mark A. Behlkei, Michael J. Welshb,c,d,g, Yi Xingb,c,f, Paul B. McCray Jr.a,b,c Author Affiliations: Department of Pediatricsa, Interdisciplinary Program in Geneticsb, Departments of Internal Medicinec, Molecular Physiology and Biophysicsd, Anatomy and Cell Biologye, Biomedical Engineeringf, Howard Hughes Medical Instituteg, Carver College of Medicine, University of Iowa, Iowa City, IA-52242 Division of Thoracic Surgeryh, Toronto General Hospital, University Health Network, University of Toronto, Toronto, Canada-M5G 2C4 Integrated DNA Technologiesi, Coralville, IA-52241 To whom correspondence should be addressed: Email: [email protected] (M.J.W.); yi- [email protected] (Y.X.); Email: [email protected] (P.B.M.) This PDF file includes: Materials and Methods References Fig. S1. miR-138 regulates SIN3A in a dose-dependent and site-specific manner. Fig. S2. miR-138 regulates endogenous SIN3A protein expression. Fig. S3. miR-138 regulates endogenous CFTR protein expression in Calu-3 cells. Fig. S4. miR-138 regulates endogenous CFTR protein expression in primary human airway epithelia. Fig. S5. miR-138 regulates CFTR expression in HeLa cells. Fig. S6. miR-138 regulates CFTR expression in HEK293T cells. Fig. S7. HeLa cells exhibit CFTR channel activity. Fig. S8. miR-138 improves CFTR processing. Fig. S9. miR-138 improves CFTR-ΔF508 processing. Fig. S10. SIN3A inhibition yields partial rescue of Cl- transport in CF epithelia. -

In Silico Tools for Splicing Defect Prediction: a Survey from the Viewpoint of End Users
© American College of Medical Genetics and Genomics REVIEW In silico tools for splicing defect prediction: a survey from the viewpoint of end users Xueqiu Jian, MPH1, Eric Boerwinkle, PhD1,2 and Xiaoming Liu, PhD1 RNA splicing is the process during which introns are excised and informaticians in relevant areas who are working on huge data sets exons are spliced. The precise recognition of splicing signals is critical may also benefit from this review. Specifically, we focus on those tools to this process, and mutations affecting splicing comprise a consider- whose primary goal is to predict the impact of mutations within the able proportion of genetic disease etiology. Analysis of RNA samples 5′ and 3′ splicing consensus regions: the algorithms used by different from the patient is the most straightforward and reliable method to tools as well as their major advantages and disadvantages are briefly detect splicing defects. However, currently, the technical limitation introduced; the formats of their input and output are summarized; prohibits its use in routine clinical practice. In silico tools that predict and the interpretation, evaluation, and prospection are also discussed. potential consequences of splicing mutations may be useful in daily Genet Med advance online publication 21 November 2013 diagnostic activities. In this review, we provide medical geneticists with some basic insights into some of the most popular in silico tools Key Words: bioinformatics; end user; in silico prediction tool; for splicing defect prediction, from the viewpoint of end users. Bio- medical genetics; splicing consensus region; splicing mutation INTRODUCTION TO PRE-mRNA SPLICING AND small nuclear ribonucleoproteins and more than 150 proteins, MUTATIONS AFFECTING SPLICING serine/arginine-rich (SR) proteins, heterogeneous nuclear ribo- Sixty years ago, the milestone discovery of the double-helix nucleoproteins, and the regulatory complex (Figure 1). -

Genetic Features of Myelodysplastic Syndrome and Aplastic Anemia in Pediatric and Young Adult Patients
Bone Marrow Failure SUPPLEMENTARY APPENDIX Genetic features of myelodysplastic syndrome and aplastic anemia in pediatric and young adult patients Siobán B. Keel, 1* Angela Scott, 2,3,4 * Marilyn Sanchez-Bonilla, 5 Phoenix A. Ho, 2,3,4 Suleyman Gulsuner, 6 Colin C. Pritchard, 7 Janis L. Abkowitz, 1 Mary-Claire King, 6 Tom Walsh, 6** and Akiko Shimamura 5** 1Department of Medicine, Division of Hematology, University of Washington, Seattle, WA; 2Clinical Research Division, Fred Hutchinson Can - cer Research Center, Seattle, WA; 3Department of Pediatric Hematology/Oncology, Seattle Children’s Hospital, WA; 4Department of Pedi - atrics, University of Washington, Seattle, WA; 5Boston Children’s Hospital, Dana Farber Cancer Institute, and Harvard Medical School, MA; 6Department of Medicine and Department of Genome Sciences, University of Washington, Seattle, WA; and 7Department of Laboratory Medicine, University of Washington, Seattle, WA, USA *SBK and ASc contributed equally to this work **TW and ASh are co-senior authors ©2016 Ferrata Storti Foundation. This is an open-access paper. doi:10.3324/haematol. 2016.149476 Received: May 16, 2016. Accepted: July 13, 2016. Pre-published: July 14, 2016. Correspondence: [email protected] or [email protected] Supplementary materials Supplementary methods Retrospective chart review Patient data were collected from medical records by two investigators blinded to the results of genetic testing. The following information was collected: date of birth, transplant, death, and last follow-up, -

Spectrum of Splicing Errors Caused by CHRNE Mutations Affecting Introns and Intron/Exon Boundaries K Ohno, a Tsujino, X-M Shen, M Milone, a G Engel
1of5 ONLINE MUTATION REPORT J Med Genet: first published as 10.1136/jmg.2004.026682 on 1 August 2005. Downloaded from Spectrum of splicing errors caused by CHRNE mutations affecting introns and intron/exon boundaries K Ohno, A Tsujino, X-M Shen, M Milone, A G Engel ............................................................................................................................... J Med Genet 2005;42:e53 (http://www.jmedgenet.com/cgi/content/full/42/8/e53). doi: 10.1136/jmg.2004.026682 Patients Background: Mutations in CHRNE, the gene encoding the Patients 1–5 (respectively a 59 year old woman, a 23 year old muscle nicotinic acetylcholine receptor e subunit, cause man, a 2.5 year old girl, a 6 year old boy, and a 44 year old congenital myasthenic syndromes. Only three of the eight man) have moderate to severe myasthenic symptoms that intronic splice site mutations of CHRNE reported to date have have been present since birth or infancy, decremental EMG had their splicing consequences characterised. responses, and no AChR antibodies. All respond partially to Methods: We analysed four previously reported and five pyridostigmine. Patient 4 underwent an intercostal muscle novel splicing mutations in CHRNE by introducing the entire biopsy for diagnosis, which showed severe endplate AChR normal and mutant genomic CHRNEs into COS cells. deficiency (6% of normal) and compensatory expression of Results and conclusions: We found that short introns (82– the fetal c-AChR at the endplate. 109 nucleotides) favour intron retention, whereas medium to long introns (306–1210 nucleotides) flanking either or both Construction of CHRNE clones for splicing analysis sides of an exon favour exon skipping. Two mutations are of To examine the consequences of the identified splice site particular interest. -

Background Splicing and Genetic Disease
Background splicing and genetic disease Diana Alexieva Imperial College London Yi Long Imperial College London Rupa Sarkar Imperial College London Hansraj Dhayan Imperial College London Emmanuel Bruet Imperial College London Robert Winston Imperial College London Igor Vorechovsky University of Southampton Leandro Castellano University of Sussex Nick Dibb ( [email protected] ) Imperial College London Research Article Keywords: background splicing, splice site mutations, cryptic splice sites, exon skipping, pseudoexons, recursive splicing, spliceosomal mutations, splicing therapy, BRCA1, BRCA1, DMD Posted Date: October 15th, 2020 DOI: https://doi.org/10.21203/rs.3.rs-92665/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 1/23 Abstract We report that low level background splicing by normal genes can be used to predict the likely effect of splicing mutations upon cryptic splice site activation and exon skipping, with emphasis on the DBASS databases, BRCA1, BRCA2 and DMD. In addition we show that background RNA splice sites are also involved in pseudoexon formation, recursive splicing and aberrant splicing in cancer. We discuss how background splicing information might inform splicing therapy. Introduction We previously established that cryptic splices sites (css) are already active, albeit at very low levels, in normal genes. We did this by using EST data to identify rare splice sites and then compared their positions to known css that are activated in human disease (1). However, this approach was limited to a minority of genes for which there was sucient EST sequence data. Since that time a large amount of RNA-sequencing data has been deposited, which we reasoned would strongly increase the power of css prediction. -

Genomic Profiling of Short- and Long-Term Caloric Restriction Effects in the Liver of Aging Mice
Genomic profiling of short- and long-term caloric restriction effects in the liver of aging mice Shelley X. Cao, Joseph M. Dhahbi, Patricia L. Mote, and Stephen R. Spindler* Department of Biochemistry, University of California, Riverside, CA 92521 Edited by Bruce N. Ames, University of California, Berkeley, CA, and approved July 11, 2001 (received for review June 19, 2001) We present genome-wide microarray expression analysis of 11,000 aging and CR on gene expression. Control young (7-month-old; n ϭ genes in an aging potentially mitotic tissue, the liver. This organ has 3) and old (27-month-old; n ϭ 3) mice were fed 95 kcal of a a major impact on health and homeostasis during aging. The effects semipurified control diet (Harlan Teklad, Madison, WI; no. of life- and health-span-extending caloric restriction (CR) on gene TD94145) per week after weaning. Long-term CR (LT-CR) young expression among young and old mice and between long-term CR (7-month-old; n ϭ 3) and old (27-month-old; n ϭ 3) mice were fed (LT-CR) and short-term CR (ST-CR) were examined. This experimental 53 kcal of a semipurified CR diet (Harlan Teklad; no. TD94146) per design allowed us to accurately distinguish the effects of aging from week after weaning. Short-term CR (ST-CR) mice were 34-month- those of CR on gene expression. Aging was accompanied by changes old control mice that were switched to 80 kcal of CR diet for 2 in gene expression associated with increased inflammation, cellular weeks, followed by 53 kcal for 2 weeks (n ϭ 3). -

Cystatin B: Mutation Detection, Alternative Splicing and Expression in Progressive Myclonus Epilepsy of Unverricht-Lundborg Type (EPM1) Patients
European Journal of Human Genetics (2007) 15, 185–193 & 2007 Nature Publishing Group All rights reserved 1018-4813/07 $30.00 www.nature.com/ejhg ARTICLE Cystatin B: mutation detection, alternative splicing and expression in progressive myclonus epilepsy of Unverricht-Lundborg type (EPM1) patients Tarja Joensuu*,1, Mervi Kuronen1, Kirsi Alakurtti1, Saara Tegelberg1, Paula Hakala1, Antti Aalto2, Laura Huopaniemi3, Nina Aula1, Roberto Michellucci4, Kai Eriksson5 and Anna-Elina Lehesjoki1 1Department of Medical Genetics and Neuroscience Center, Folkha¨lsan Institute of Genetics, Biomedicum Helsinki, University of Helsinki, Finland; 2Institute of Biotechnology, Viikki Biocenter, University of Helsinki, Finland; 3Rational Drug Design Program, Biomedicum Helsinki, Helsinki, Finland; 4Department of Neurosciences, Epilepsy Centre, Bellaria Hospital, Bologna, Italy; 5Pediatric Neurology Unit, Department of Pediatrics, Pediatric Research Center, Medical School, University of Tampere and Tampere University Hospital, Tampere, Finland Progressive myoclonus epilepsy of Unverricht-Lundborg type (EPM1) is an autosomal recessive neurodegenerative disorder caused by mutations in the cystatin B gene (CSTB) that encodes an inhibitor of several lysosomal cathepsins. An unstable expansion of a dodecamer repeat in the CSTB promoter accounts for the majority of EPM1 disease alleles worldwide. We here describe a novel PCR protocol for detection of the dodecamer repeat expansion. We describe two novel EPM1-associated mutations, c.149G4A leading to the p.G50E missense change and an intronic 18-bp deletion (c.168 þ 1_18del), which affects splicing of CSTB. The p.G50E mutation that affects the conserved QVVAG amino acid sequence critical for cathepsin binding fails to associate with lysosomes. This further supports the previously implicated physiological importance of the CSTB-lysosome association.