University Microfilms, Inc., Ann Arbor, Michigan OXIDIZING PROPERTIES of SELENIUM COMPOUNDS
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Retention Indices for Frequently Reported Compounds of Plant Essential Oils
Retention Indices for Frequently Reported Compounds of Plant Essential Oils V. I. Babushok,a) P. J. Linstrom, and I. G. Zenkevichb) National Institute of Standards and Technology, Gaithersburg, Maryland 20899, USA (Received 1 August 2011; accepted 27 September 2011; published online 29 November 2011) Gas chromatographic retention indices were evaluated for 505 frequently reported plant essential oil components using a large retention index database. Retention data are presented for three types of commonly used stationary phases: dimethyl silicone (nonpolar), dimethyl sili- cone with 5% phenyl groups (slightly polar), and polyethylene glycol (polar) stationary phases. The evaluations are based on the treatment of multiple measurements with the number of data records ranging from about 5 to 800 per compound. Data analysis was limited to temperature programmed conditions. The data reported include the average and median values of retention index with standard deviations and confidence intervals. VC 2011 by the U.S. Secretary of Commerce on behalf of the United States. All rights reserved. [doi:10.1063/1.3653552] Key words: essential oils; gas chromatography; Kova´ts indices; linear indices; retention indices; identification; flavor; olfaction. CONTENTS 1. Introduction The practical applications of plant essential oils are very 1. Introduction................................ 1 diverse. They are used for the production of food, drugs, per- fumes, aromatherapy, and many other applications.1–4 The 2. Retention Indices ........................... 2 need for identification of essential oil components ranges 3. Retention Data Presentation and Discussion . 2 from product quality control to basic research. The identifi- 4. Summary.................................. 45 cation of unknown compounds remains a complex problem, in spite of great progress made in analytical techniques over 5. -

Selenium Stabilized Carbanions. Preparation of A-Lithio Selenides and Applications to the Synthesis of Olefins by Reductive Elim
6638 Journal of the American Chemical Society / 101:22 / October 24, I979 Selenium Stabilized Carbanions. Preparation of a-Lithio Selenides and Applications to the Synthesis of Olefins by Reductive Elimination of ,&Hydroxy Selenides and Selenoxide Syn Elimination Hans J. Reich,** Flora Chow, and Shrenik K. Shah Contribution from the Department of Chemistry, University of Wisconsin-Madison, Madison, Wisconsin 53706. Received May 10, 1979 Abstract: The deprotonation of several alkyl aryl selenides with lithium amide bases has been studied. The kinetic acidity of methyl m-trifluoromethylphenyl selenide was found to be 1/33 that of the sulfur analogue. The introduction of a m-trifluoro- methyl substituent into methyl phenyl sulfide increased the kinetic acidity by a factor of 22.4. A variety of @-hydroxy selenides have been prepared by the reduction of a-phenylseleno ketones and the addition of a-lithio selenides (prepared by deprotona- tion of benzyl phenyl selenide, bis(phenylseleno)methane, methoxymethyl m-trifluoromethylphenyl selenide, and phenylsele- noacetic acid, and by n-butyllithium cleavage of bis(pheny1seleno)methane) to carbonyl compounds. These @-hydroxyselen- ides are converted to olefins on treatment with rnethanesulfonyl chloride and triethylamine. This reductive elimination pro- ceeds exclusively or predominantly with anti stereochemistry. Simple olefins, styrenes, cinnamic acids, vinyl ethers, and vinyl selenides have been prepared using this technique. Attempts to carry out the syn reductive elimination of @-hydroxyselenox- -

United States Patent (19) 11, 3,989,755 Mccoy Et Al
United States Patent (19) 11, 3,989,755 McCoy et al. (45) Nov. 2, 1976 54 PRODUCTION OF OXIMES BY THE (56) References Cited REACTION OF CARBON MONOXDE WITH UNITED STATES PATENTS NTROCOMPOUNDS 2,945,065 7/1960 Donaruma...................... 260/566 A 75) Inventors: John J. McCoy, Media; John G. 3,480,672 11/1969 Kober et al..................... 260/566 A Zajacek, Strafford, both of Pa.; Karl 3,734,964 5/1973 Knifton........................... 260/566. A E. Fuger, Therwil, Switzerland (73) Assignee: Atlantic Richfield Company, Los Primary Examiner-Gerald A. Schwartz Angeles, Calif. Attorney, Agent, or Firm-Delbert E. McCaslin 22 Filed: Feb. 20, 1975 (21) Appl. No.: 551,487 57) ABSTRACT Related U.S. Application Data Production of oximes (and ketones) by contacting at 63) Continuation-in-part of Ser. No. 372,457, June 21, elevated temperatures and pressures, a primary or sec 1973, abandoned. ondary saturated aliphatic nitrocompound with carbon monoxide in the presence of a catalyst comprising me 52 U.S. C. ........................ 260/566 A; 260/586 C; tallic selenium or inorganic selenium compounds and 260/586 R; 260/593 R a base. (51 int. Cl”........................................ C07C 131/04 58) Field of Search........ 260/566 A, 586 A, 586 R, 20 Claims, No Drawings 260/593 R 3,989,755 2 cycloaliphatic nitrocompound is contacted with carbon PRODUCTION OF OXIMES BY THE REACTION OF monoxide at temperatures in the range of from 50° to 200 C. under pressures in the range of from 10 atmo CARBON MONOXIDE WITH NITROCOMPOUNDS spheres to 200 atmospheres in the presence of a sele nium catalyst and a base. -
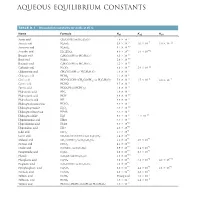
Aqueous Equilibrium Constants
BLB_APP_D_1062-1063hr.qxp 11/8/10 2:27 PM Page 1062 APPENDIX D AQUEOUS EQUILIBRIUM CONSTANTS TABLE D.1 • Dissociation Constants for Acids at 25 ˚C Name Formula Ka1 Ka2 Ka3 -5 Acetic acid CH3COOH (or HC2H3O2) 1.8 * 10 * -3 * -7 * -12 Arsenic acid H3AsO4 5.6 10 1.0 10 3.0 10 * -10 Arsenous acid H3AsO3 5.1 10 * -5 * -12 Ascorbic acid H2C6H6O6 8.0 10 1.6 10 * -5 Benzoic acid C6H5COOH (or HC7H5O2) 6.3 10 * -10 Boric acid H3BO3 5.8 10 * -5 Butanoic acid C3H7COOH (or HC4H7O2) 1.5 10 * -7 * -11 Carbonic acid H2CO3 4.3 10 5.6 10 * -3 Chloroacetic acid CH2ClCOOH (or HC2H2O2Cl) 1.4 10 * -2 Chlorous acid HClO2 1.1 10 * -4 * -5 -7 Citric acid HOOCC(OH) (CH2COOH)2 (or H3C6H5O7) 7.4 10 1.7 10 4.0 * 10 - Cyanic acid HCNO 3.5 * 10 4 * -4 Formic acid HCOOH (or HCHO2) 1.8 10 * -5 Hydroazoic acid HN3 1.9 10 - Hydrocyanic acid HCN 4.9 * 10 10 - Hydrofluoric acid HF 6.8 * 10 4 - -7 Hydrogen chromate ion HCrO4 3.0 * 10 * -12 Hydrogen peroxide H2O2 2.4 10 - -2 Hydrogen selenate ion HSeO4 2.2 * 10 * -8 * -19 Hydrogen sulfide H2S 9.5 10 1 10 - Hypobromous acid HBrO 2.5 * 10 9 - Hypochlorous acid HClO 3.0 * 10 8 - Hypoiodous acid HIO 2.3 * 10 11 * -1 Iodic acid HIO3 1.7 10 * -4 Lactic acid CH3CH(OH)COOH (or HC3H5O3) 1.4 10 * -3 * -6 Malonic acid CH2(COOH)2 (or H2C3H2O4) 1.5 10 2.0 10 * -4 Nitrous acid HNO2 4.5 10 * -2 * -5 Oxalic acid (COOH)2 (or H2C2O4) 5.9 10 6.4 10 * -2 * -9 Paraperiodic acid H5IO6 2.8 10 5.3 10 * -10 Phenol C6H5OH (or HC6H5O) 1.3 10 * -3 * -8 * -13 Phosphoric acid H3PO4 7.5 10 6.2 10 4.2 10 * -5 Propionic acid C2H5COOH (or HC3H5O2) 1.3 10 * -

Zn-Nx Sites on N-Doped Carbon for Aerobic Oxidative Cleavage
ARTICLE https://doi.org/10.1038/s41467-021-25118-0 OPEN Zn-Nx sites on N-doped carbon for aerobic oxidative cleavage and esterification of C(CO)-C bonds ✉ ✉ Chao Xie1, Longfei Lin 2, Liang Huang 3, Zixin Wang1, Zhiwei Jiang 1, Zehui Zhang1 & Buxing Han 2 Selective cleavage of C-C bonds is very important in organic chemistry, but remains chal- lenging because of their inert chemical nature. Herein, we report that Zn/NC-X catalysts, in 1234567890():,; which Zn2+ coordinate with N species on microporous N-doped carbon (NC) and X denotes the pyrolysis temperature, can effectively catalyze aerobic oxidative cleavage of C(CO)-C bonds and quantitatively convert acetophenone to methyl benzoate with a yield of 99% at 100 °C. The Zn/NC-950 can be applied for a wide scope of acetophenone derivatives as well as more challenging alkyl ketones. Detail mechanistic investigations reveal that the catalytic performance of Zn/NC-950 can be attributed to the coordination between Zn2+ and N species to change the electronic state of the metal, synergetic effect of the Zn single sites with their surrounding N atoms, as well as the microporous structure with the high surface area and structural defects of the NC. 1 Key Laboratory of Catalysis and Energy Materials Chemistry of Ministry of Education & Hubei Key Laboratory of Catalysis and Materials Science, South- Central University for Nationalities, Wuhan, China. 2 Beijing National Laboratory for Molecular Sciences, CAS Key Laboratory of Colloid, Interface and Chemical Thermodynamics, Institute of Chemistry, Chinese Academy of Sciences, Beijing, China. 3 The State Key Laboratory of Refractories and Metallurgy, ✉ Wuhan University of Science and Technology, Wuhan, China. -

Synthesis and Consecutive Reactions of Α-Azido Ketones: a Review
Molecules 2015, 20, 14699-14745; doi:10.3390/molecules200814699 OPEN ACCESS molecules ISSN 1420-3049 www.mdpi.com/journal/molecules Review Synthesis and Consecutive Reactions of α-Azido Ketones: A Review Sadia Faiz 1,†, Ameer Fawad Zahoor 1,*, Nasir Rasool 1,†, Muhammad Yousaf 1,†, Asim Mansha 1,†, Muhammad Zia-Ul-Haq 2,† and Hawa Z. E. Jaafar 3,* 1 Department of Chemistry, Government College University Faisalabad, Faisalabad-38000, Pakistan, E-Mails: [email protected] (S.F.); [email protected] (N.R.); [email protected] (M.Y.); [email protected] (A.M.) 2 Office of Research, Innovation and Commercialization, Lahore College for Women University, Lahore-54600, Pakistan; E-Mail: [email protected] 3 Department of Crop Science, Faculty of Agriculture, Universiti Putra Malaysia, Serdang-43400, Selangor, Malaysia † These authors contributed equally to this work. * Authors to whom correspondence should be addressed; E-Mails: [email protected] (A.F.Z.); [email protected] (H.Z.E.J.); Tel.: +92-333-6729186 (A.F.Z.); Fax: +92-41-9201032 (A.F.Z.). Academic Editors: Richard A. Bunce, Philippe Belmont and Wim Dehaen Received: 20 April 2015 / Accepted: 3 June 2015 / Published: 13 August 2015 Abstract: This review paper covers the major synthetic approaches attempted towards the synthesis of α-azido ketones, as well as the synthetic applications/consecutive reactions of α-azido ketones. Keywords: α-azido ketones; synthetic applications; heterocycles; click reactions; drugs; azides 1. Introduction α-Azido ketones are very versatile and valuable synthetic intermediates, known for their wide variety of applications, such as in amine, imine, oxazole, pyrazole, triazole, pyrimidine, pyrazine, and amide alkaloid formation, etc. -

Developing a Method for Determining the Mass Balance of Selenium and Tellurium Bioprocessed by a Selenium-Resistant Bacterium Grown in The
Developing a Method for Determining the Mass Balance of Selenium and Tellurium Bioprocessed by a Selenium-Resistant Bacterium Grown in the Presence of Selenite or Tellurite __________________ A Thesis Presented to The Faculty of the Department of Chemistry Sam Houston State University ____________________ In Partial Fulfillment Of the Requirements for the Degree of Master of Science ____________________ by Janet Horton Bius December, 2001 Developing a Method for Determining the Mass Balance of Selenium and Tellurium Bioprocessed by a Selenium-Resistant Bacterium Grown in the Presence of Selenite or Tellurite by Janet Horton Bius ____________________ Approved: _____________________________ Thomas G. Chasteen ____________________________ Mary F. Plishker ____________________________ Rick C. White Approved: ____________________________ Brian Chapman, Dean College of Arts and Sciences II ABSTRACT Bius, Janet Horton, Developing a Method for Determining the Mass Balance of Selenium and Tellurium Bioprocessed by a Selenium-Resistant Bacterium Grown in the Presence of Selenite or Tellurite, Master of Science (Chemistry), December, 2001. Sam Houston State University, Hunts- ville, Texas, 68 pp. Purpose The purpose of this investigation was to determine: (1) the mass balance of selenium or tellurium that was bioreduced when a selenium-resistant facultative anaerobe was amended with either selenium or tellurium; and (2) methods to analyze for these metalloids in biological samples. Methods Analytical methods were developed for the determination of -

Working with Hazardous Chemicals
A Publication of Reliable Methods for the Preparation of Organic Compounds Working with Hazardous Chemicals The procedures in Organic Syntheses are intended for use only by persons with proper training in experimental organic chemistry. All hazardous materials should be handled using the standard procedures for work with chemicals described in references such as "Prudent Practices in the Laboratory" (The National Academies Press, Washington, D.C., 2011; the full text can be accessed free of charge at http://www.nap.edu/catalog.php?record_id=12654). All chemical waste should be disposed of in accordance with local regulations. For general guidelines for the management of chemical waste, see Chapter 8 of Prudent Practices. In some articles in Organic Syntheses, chemical-specific hazards are highlighted in red “Caution Notes” within a procedure. It is important to recognize that the absence of a caution note does not imply that no significant hazards are associated with the chemicals involved in that procedure. Prior to performing a reaction, a thorough risk assessment should be carried out that includes a review of the potential hazards associated with each chemical and experimental operation on the scale that is planned for the procedure. Guidelines for carrying out a risk assessment and for analyzing the hazards associated with chemicals can be found in Chapter 4 of Prudent Practices. The procedures described in Organic Syntheses are provided as published and are conducted at one's own risk. Organic Syntheses, Inc., its Editors, and its Board of Directors do not warrant or guarantee the safety of individuals using these procedures and hereby disclaim any liability for any injuries or damages claimed to have resulted from or related in any way to the procedures herein. -

Chemical Chemical Hazard and Compatibility Information
Chemical Chemical Hazard and Compatibility Information Acetic Acid HAZARDS & STORAGE: Corrosive and combustible liquid. Serious health hazard. Reacts with oxidizing and alkali materials. Keep above freezing point (62 degrees F) to avoid rupture of carboys and glass containers.. INCOMPATIBILITIES: 2-amino-ethanol, Acetaldehyde, Acetic anhydride, Acids, Alcohol, Amines, 2-Amino-ethanol, Ammonia, Ammonium nitrate, 5-Azidotetrazole, Bases, Bromine pentafluoride, Caustics (strong), Chlorosulfonic acid, Chromic Acid, Chromium trioxide, Chlorine trifluoride, Ethylene imine, Ethylene glycol, Ethylene diamine, Hydrogen cyanide, Hydrogen peroxide, Hydrogen sulfide, Hydroxyl compounds, Ketones, Nitric Acid, Oleum, Oxidizers (strong), P(OCN)3, Perchloric acid, Permanganates, Peroxides, Phenols, Phosphorus isocyanate, Phosphorus trichloride, Potassium hydroxide, Potassium permanganate, Potassium-tert-butoxide, Sodium hydroxide, Sodium peroxide, Sulfuric acid, n-Xylene. Acetone HAZARDS & STORAGE: Store in a cool, dry, well ventilated place. INCOMPATIBILITIES: Acids, Bromine trifluoride, Bromine, Bromoform, Carbon, Chloroform, Chromium oxide, Chromium trioxide, Chromyl chloride, Dioxygen difluoride, Fluorine oxide, Hydrogen peroxide, 2-Methyl-1,2-butadiene, NaOBr, Nitric acid, Nitrosyl chloride, Nitrosyl perchlorate, Nitryl perchlorate, NOCl, Oxidizing materials, Permonosulfuric acid, Peroxomonosulfuric acid, Potassium-tert-butoxide, Sulfur dichloride, Sulfuric acid, thio-Diglycol, Thiotrithiazyl perchlorate, Trichloromelamine, 2,4,6-Trichloro-1,3,5-triazine -

Carboligation Using the Aldol Reaction
Digital Comprehensive Summaries of Uppsala Dissertations from the Faculty of Science and Technology 1730 Carboligation using the aldol reaction A comparison of stereoselectivity and methods DERAR AL-SMADI ACTA UNIVERSITATIS UPSALIENSIS ISSN 1651-6214 ISBN 978-91-513-0472-4 UPPSALA urn:nbn:se:uu:diva-362866 2018 Dissertation presented at Uppsala University to be publicly examined in BMC C2:301, Husargatan 3, Uppsala, Friday, 30 November 2018 at 09:15 for the degree of Doctor of Philosophy. The examination will be conducted in English. Faculty examiner: Professor Ulf Nilsson (Lund University). Abstract Al-Smadi, D. 2018. Carboligation using the aldol reaction. A comparison of stereoselectivity and methods. Digital Comprehensive Summaries of Uppsala Dissertations from the Faculty of Science and Technology 1730. 50 pp. Uppsala: Acta Universitatis Upsaliensis. ISBN 978-91-513-0472-4. The research summarized in this thesis focuses on synthesizing aldehyde and aldol compounds as substrates and products for the enzyme D-fructose-6-aldolase (FSA). Aldolases are important enzymes for the formation of carbon-carbon bonds in nature. In biological systems, aldol reactions, both cleavage and formation play central roles in sugar metabolism. Aldolases exhibit high degrees of stereoselectivity and can steer the product configurations to a given enantiomeric and diastereomeric form. To become truly useful synthetic tools, the substrate scope of these enzymes needs to become broadened. In the first project, phenylacetaldehyde derivatives were synthesized for the use as test substrates for E. coli FSA. Different methods were discussed to prepare phenylacetaldehyde derivatives, the addition of a one carbon unit to benzaldehyde derivatives using a homologation reaction was successful and was proven efficient and non-sensitive to the moisture. -

Is Selenium a Metal, Non-Metal Or Metalloid?
Is Selenium a metal, non-metal or metalloid? Abstract Is selenium(Se) a metal, non-metal, or a metalloid? There are various public opinions circulating around it. Since a long time from now, there are a lot of voices discussing this. Even until now, there is still no consensus about it. So, in this project, we are trying to find out whether selenium is a non-metal, metal or metalloids base on its physical and chemical properties which could be studied in the secondary school combining with the other information from the internet. Principles and hypothesis Studied from the secondary school chemistry, the general properties of metals include being good conductors of heat and electricity, having high melting and boiling points. Non-metals generally have a lower melting point and boiling point than metals and they being poor conductors of heat and electricity, etc. And the physical properties of metalloids are having extremely high melting/ boiling point, and having fair electrical conductivity. On the other hand, the oxides of metal are generally basic whereas the oxide of non-metals and metalloids are generally acidic. We will define selenium’s chemical category based on the above properties. Besides, we would like to introduce the concept of displacement reaction in studying the chemical properties of the chalcogens(e.g. sulphur(S) and selenium(Se)), especially selenium such rarely mentioned element. We can assume selenium is a metal if selenium could displace a metal oxide or a metal could displace selenium (IV) oxide. If selenium is a non-metal, its oxide could be displaced by sulphur which is supposed to be more reactive than selenium in the chalcogen group. -

Why Nature Chose Selenium Hans J
Reviews pubs.acs.org/acschemicalbiology Why Nature Chose Selenium Hans J. Reich*, ‡ and Robert J. Hondal*,† † University of Vermont, Department of Biochemistry, 89 Beaumont Ave, Given Laboratory, Room B413, Burlington, Vermont 05405, United States ‡ University of WisconsinMadison, Department of Chemistry, 1101 University Avenue, Madison, Wisconsin 53706, United States ABSTRACT: The authors were asked by the Editors of ACS Chemical Biology to write an article titled “Why Nature Chose Selenium” for the occasion of the upcoming bicentennial of the discovery of selenium by the Swedish chemist Jöns Jacob Berzelius in 1817 and styled after the famous work of Frank Westheimer on the biological chemistry of phosphate [Westheimer, F. H. (1987) Why Nature Chose Phosphates, Science 235, 1173−1178]. This work gives a history of the important discoveries of the biological processes that selenium participates in, and a point-by-point comparison of the chemistry of selenium with the atom it replaces in biology, sulfur. This analysis shows that redox chemistry is the largest chemical difference between the two chalcogens. This difference is very large for both one-electron and two-electron redox reactions. Much of this difference is due to the inability of selenium to form π bonds of all types. The outer valence electrons of selenium are also more loosely held than those of sulfur. As a result, selenium is a better nucleophile and will react with reactive oxygen species faster than sulfur, but the resulting lack of π-bond character in the Se−O bond means that the Se-oxide can be much more readily reduced in comparison to S-oxides.