Synthetic Applications of Intramolecular Thiol-Ene “Click” Reactions
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

8.6 Acidity of Alcohols and Thiols 355
08_BRCLoudon_pgs5-1.qxd 12/8/08 11:05 AM Page 355 8.6 ACIDITY OF ALCOHOLS AND THIOLS 355 ural barrier to the passage of ions. However, the hydrocarbon surface of nonactin allows it to enter readily into, and pass through, membranes. Because nonactin binds and thus transports ions, the ion balance crucial to proper cell function is upset, and the cell dies. Ion Channels Ion channels, or “ion gates,” provide passageways for ions into and out of cells. (Recall that ions are not soluble in membrane phospholipids.) The flow of ions is essen- tial for the transmission of nerve impulses and for other biological processes. A typical chan- nel is a large protein molecule imbedded in a cell membrane. Through various mechanisms, ion channels can be opened or closed to regulate the concentration of ions in the interior of the cell. Ions do not diffuse passively through an open channel; rather, an open channel contains regions that bind a specific ion. Such an ion is bound specifically within the channel at one side of the membrane and is somehow expelled from the channel on the other side. Remark- ably, the structures of the ion-binding regions of these channels have much in common with the structures of ionophores such as nonactin. The first X-ray crystal structure of a potassium- ion channel was determined in 1998 by a team of scientists at Rockefeller University led by Prof. Roderick MacKinnon (b. 1956), who shared the 2003 Nobel Prize in Chemistry for this work. The interior of the channel contains binding sites for two potassium ions; these sites are oxygen-rich, much like the interior of nonactin. -

THIOL OXIDATION a Slippery Slope the Oxidation of Thiols — Molecules RSH Oxidation May Proceed Too Predominates
RESEARCH HIGHLIGHTS Nature Reviews Chemistry | Published online 25 Jan 2017; doi:10.1038/s41570-016-0013 THIOL OXIDATION A slippery slope The oxidation of thiols — molecules RSH oxidation may proceed too predominates. Here, the maximum of the form RSH — can afford quickly for intermediates like RSOH rate constants indicate the order − − − These are many products. From least to most to be spotted and may also afford of reactivity: RSO > RS >> RSO2 . common oxidized, these include disulfides intractable mixtures. Addressing When the reactions are carried out (RSSR), as well as sulfenic (RSOH), the first problem, Chauvin and Pratt in methanol-d , the obtained kinetic reactions, 1 sulfinic (RSO2H) and sulfonic slowed the reactions down by using isotope effect values (kH/kD) are all but have (RSO3H) acids. Such chemistry “very sterically bulky thiols, whose in the range 1.1–1.2, indicating that historically is pervasive in nature, in which corresponding sulfenic acids were no acidic proton is transferred in the been very disulfide bonds between cysteine known to be isolable but were yet rate-determining step. Rather, the residues stabilize protein structures, to be thoroughly explored in terms oxidations involve a specific base- difficult to and where thiols and thiolates often of reactivity”. The second problem catalysed mechanism wherein an study undergo oxidation by H2O2 or O2 in was tackled by modifying the model acid–base equilibrium precedes the order to protect important biological system, 9-mercaptotriptycene, by rate-determining nucleophilic attack − − − structures from damage. Among including a fluorine substituent to of RS , RSO or RSO2 on H2O2. the oxidation products, sulfenic serve as a spectroscopic handle. -

Thiolated Polymers: Stability of Thiol Moieties Under Different Storage Conditions
Scientia Pharmaceutica (Sci. Pharm.) 70, 331-339 (2002) 0 Osterreichische Apotheker-Verlagsgesellschaftm.b.H., Wien, Printed in Austria Thiolated polymers: Stability of thiol moieties under different storage conditions A. ~ernko~-schnijrchi*,M.D. ~ornof'*,C.E. ~ast'and N. ~angoth' 'institute of Pharmaceutical Technology and Biopharmaceutics, Center of Pharmacy, University of Vienna, Althanstr. 14, 1090 Vienna, Austria 2~romaPharma GmbH, lndustriezeile 6, 2100 Leobendorf, Austria Abstract: The purpose of this study was to evaluate the stability of thiolated polymers - so-called thiomers. A polycarbophil-cysteine conjugate and a chitosan-thioglycolic acid conjugate were chosen as representative anionic and cationic thiomer. The thiol group bearing compounds L-cysteine and thioglycolic acid were introduced to polycarbophil and chitosan, respectively with a coupling reaction mediated by a carbodiimide. The resulting thiolated polymers were freeze-dried and the amount of thiol groups on the thiomer was determined spectrophotometrically. Each kind of polymer was directly used or compressed into 1 mg matrix-tablets. Polymers were stored for a period of six months at four different storage conditions, namely at -20°C (56% relative humidity; RH), 4°C (53% RH), at 20°C (70% RH), and at 22°C (25% RH). Samples were taken after 6 months to determine the formation of disulfide bonds and the remaining thiol groups on the polymer. When the polycarbophil-cysteine and chitosan-thioglycolic acid conjugate were stored as powder a decrease of free thiol groups was observed only after storage at 20°C and 70% RH. Both polymers were found to be stable under all storage conditions when compressed into matrix tablets. -

In This Handout, All of Our Functional Groups Are Presented As Condensed Line Formulas, 2D and 3D Formulas and with Nomenclature Prefixes and Suffixes (If Present)
In this handout, all of our functional groups are presented as condensed line formulas, 2D and 3D formulas and with nomenclature prefixes and suffixes (if present). Organic names are built on a foundation of alkanes, alkenes and alkynes. Those examples are presented first and you need to know those rules. The strategies can be found in Chapter 4 of our textbook (alkanes: pages 93-98, cycloalkanes 102-104, alkenes: pages 104-110, alkynes: pages 112-113 and combinations of all of them 113-115). After introducing examples of alkanes, alkenes, alkynes and combinations of them, the functional groups are presented in order of priority. A few nomenclature examples are provided for each of the functional groups. Examples of the various functional groups are presented on pages 115-135 in the textbook. Two overview pages are on pages 136-137. Some functional groups have a suffix name when they are the highest priority functional group and a prefix name when they are not the highest priority group, and these are added to the skeletal names with identifying numbers and stereochemistry terms (E and Z for alkenes, R and S for chiral centers and cis and trans for rings). Several low priority functional groups only have a prefix name. A few additional special patterns are shown on pages 98-102. The only way to learn this topic is practice (over and over). The best practice approach is to actually write out the names (on an extra piece of paper or on a white board, and then do it again). The same functional groups are used throughout the entire course. -

Naming Ethers and Thiols (Naming and Properties)
Ethers • Contain an ─O─ between two carbon groups. • That are simple are named by listing the alkyl names in alphabetical order followed by ether Cyclic ethers (heterocyclic compounds) are often given common names. DO NOT memorize! IUPAC Names for Ethers The shorter alkyl group and the oxygen are named as an alkoxy group attached to the longer hydrocarbon. methoxy propane CH3—O—CH2—CH2—CH3 1 2 3 Numbering the longer alkane gives: 1-methoxypropane Treat all R-O- groups that you find in a molecule as alkoxy groups or branches (substituents), and follow the previous rules we’ve covered for all other families. Properties of Ethers • Slightly polar. • Cannot be H-bond donors. • Are H-bond acceptors (soluble in water up to 4 C’s). • Simple ethers are highly flammable (many form explosive peroxides). Anesthetics • Inhibit pain signals to the brain. • Such as ethyl ether CH3─CH2─O─CH2─CH3 were used for over a century, but caused nausea and were flammable. • Developed by 1960s were nonflammable. Cl F F Cl F H │ │ │ │ │ │ H─C─C─O─C─H H─C─C─O─C─H │ │ │ │ │ │ F F F H F H Ethane(enflurane) Penthrane Methyl tert-butyl ether (MTBE) • Is one of the most produced organic chemicals. • Is a fuel additive used to improve gasoline combustion. • Use is questioned since the discovery that MTBE has contaminated water supplies. Reyes Free to copy for educational purposes Thiols Thiols or mercaptans, are sulfur analogs of alcohols. (The –SH group is called the mercapto, or sulfhydryl group.) The IUPAC nomenclature system adds the ending –thiol to the name of the alkane, but without dropping the final –e. -

Interplay of Hydrophobic Thiol and Polar Epoxy Silicate Groups on Microstructural Development in Low-Alcohol, Crosslinked Sol–Gel Coatings for Corrosion Prevention
coatings Article Interplay of Hydrophobic Thiol and Polar Epoxy Silicate Groups on Microstructural Development in Low-Alcohol, Crosslinked Sol–Gel Coatings for Corrosion Prevention Shegufa Shetranjiwalla * , Andrew J. Vreugdenhil and Oliver Strong Department of Chemistry, Inorganic Materials Research Laboratory, Trent University, 1600 West Bank Drive, Peterborough, ON K9L 0G2, Canada; [email protected] (A.J.V.); [email protected] (O.S.) * Correspondence: [email protected]; Tel.: +1-705-748-1011 (ext. 7319) Abstract: We have demonstrated that our patented, crosslinked, sol–gel, epoxy–thiol silicates made from the combination of (a) tetraethoxysilane (TEOS, T), 3-glycidoxypropyltrimethoxysilane (GPTMS, G), and the (b) sulfur-containing 3-mercaptopropyltrimethoxysilane (MPTMS, S) with TEOS in a 1:1 stoichiometric ratio form the 1:1 TGST (crosslinked epoxy and thiol silicates) coating, which can be successfully utilized for the corrosion protection of low-carbon steel. Alcohols that are a by-product of sol–gel reactions influence the network formation, crosslinking density, and formulation stability, are volatile organic contents, and are regulated in the coatings industry. To improve environmental sustainability, a series of low-alcohol (LA) formulations with TG:ST ratios of 3:1 to 1:3 was prepared to investigate the microstructural development and crosslinking reactions emerging from the interplay of the hydrophobic thiol and polar epoxy silicates induced by the low-alcohol environment. The Citation: Shetranjiwalla, S.; impact on crosslinking density was characterized by Fourier Transform Infrared (FTIR), Raman, XPS, Vreugdenhil, A.J.; Strong, O. Interplay viscosity, and pot-life measurements. Low-alcohol TGST (LA(TGST)) formulations were compared, of Hydrophobic Thiol and Polar Epoxy Silicate Groups on using the example of 1:1 TGST, to corresponding TGST formulations where alcohols were retained. -

Catalysis of Peroxide Reduction by Fast Reacting Protein Thiols Focus Review †,‡ †,‡ ‡,§ ‡,§ ∥ Ari Zeida, Madia Trujillo, Gerardo Ferrer-Sueta, Ana Denicola, Darío A
Review Cite This: Chem. Rev. 2019, 119, 10829−10855 pubs.acs.org/CR Catalysis of Peroxide Reduction by Fast Reacting Protein Thiols Focus Review †,‡ †,‡ ‡,§ ‡,§ ∥ Ari Zeida, Madia Trujillo, Gerardo Ferrer-Sueta, Ana Denicola, Darío A. Estrin, and Rafael Radi*,†,‡ † ‡ § Departamento de Bioquímica, Centro de Investigaciones Biomedicaś (CEINBIO), Facultad de Medicina, and Laboratorio de Fisicoquímica Biologica,́ Facultad de Ciencias, Universidad de la Republica,́ 11800 Montevideo, Uruguay ∥ Departamento de Química Inorganica,́ Analítica y Química-Física and INQUIMAE-CONICET, Facultad de Ciencias Exactas y Naturales, Universidad de Buenos Aires, 2160 Buenos Aires, Argentina ABSTRACT: Life on Earth evolved in the presence of hydrogen peroxide, and other peroxides also emerged before and with the rise of aerobic metabolism. They were considered only as toxic byproducts for many years. Nowadays, peroxides are also regarded as metabolic products that play essential physiological cellular roles. Organisms have developed efficient mechanisms to metabolize peroxides, mostly based on two kinds of redox chemistry, catalases/peroxidases that depend on the heme prosthetic group to afford peroxide reduction and thiol-based peroxidases that support their redox activities on specialized fast reacting cysteine/selenocysteine (Cys/Sec) residues. Among the last group, glutathione peroxidases (GPxs) and peroxiredoxins (Prxs) are the most widespread and abundant families, and they are the leitmotif of this review. After presenting the properties and roles of different peroxides in biology, we discuss the chemical mechanisms of peroxide reduction by low molecular weight thiols, Prxs, GPxs, and other thiol-based peroxidases. Special attention is paid to the catalytic properties of Prxs and also to the importance and comparative outlook of the properties of Sec and its role in GPxs. -

Thiol–Ene Click Reaction Initiated Rapid Gelation of PEGDA/Silk Fibroin Hydrogels
polymers Article Thiol–Ene Click Reaction Initiated Rapid Gelation of PEGDA/Silk Fibroin Hydrogels Jianwei Liang, Xiaoning Zhang * , Zhenyu Chen, Shan Li and Chi Yan State Key Laboratory of Silkworm Genome Biology, College of Biotechnology, Southwest University, Chongqing 400715, China; [email protected] (J.L.); [email protected] (Z.C.); [email protected] (S.L.); [email protected] (C.Y.) * Correspondence: [email protected]; Tel.: +86-1592-281-5612 Received: 7 October 2019; Accepted: 12 December 2019; Published: 14 December 2019 Abstract: In this work, poly(ethylene glycol) diacrylate (PEGDA) molecules were grafted to silk fibroin (SF) molecules via a thiol–ene click reaction under 405 nm UV illumination for the fabrication of a PEGDA/SF composite hydrogel. The composite hydrogels could be prepared in a short and controllable gelation time without the use of a photoinitiator. Features relevant to the drug delivery of the PEGDA/SF hydrogels were assessed, and the hydrogels were characterized by various techniques. The results showed that the prepared PEGDA/SF hydrogels demonstrated a good sustained-release performance with limited swelling behavior. It was found that a prior cooling step can improve the compressive strength of the hydrogels effectively. Additionally, the MTT assay indicated the prepared PEGDA/SF hydrogel is non-cytotoxic. Subcutaneous implantation of the PEGDA/SF hydrogel in Kunming mice did not induce an obvious inflammation, which revealed that the prepared PEGDA/SF hydrogel possessed good biocompatibility. Furthermore, the mechanism of the gelation process was discussed. Keywords: silk fibroin; PEGDA; thiol–ene click reaction; gelation; drug delivery 1. Introduction It is well known that qualified biomaterials shall possess good mechanical properties and biological compatibility [1]. -

Thiol–Ene Photopolymerization: Scaling Law and Analytical Formulas for Conversion Based on Kinetic Rate and Thiol–Ene Molar Ratio
polymers Article Thiol–Ene Photopolymerization: Scaling Law and Analytical Formulas for Conversion Based on Kinetic Rate and Thiol–Ene Molar Ratio Kuo-Ti Chen 1, Da-Chuan Cheng 2, Jui-Teng Lin 3 and Hsia-Wei Liu 4,* 1 Graduate Institute of Applied Science and Engineering, Fu Jen Catholic University, New Taipei City 24205, Taiwan; [email protected] 2 Department of Biomedical Imaging and Radiological Science, China Medical University, Taizhong 400, Taiwan; [email protected] 3 New Vision, Inc., New Taipei City 242, Taiwan; [email protected] 4 Department of Life Science, Fu Jen Catholic University, New Taipei City 24205, Taiwan * Correspondence: [email protected]; Tel.: +886-0910-688-968 Received: 22 September 2019; Accepted: 7 October 2019; Published: 10 October 2019 Abstract: Kinetics and analytical formulas for radical-mediated thiol–ene photopolymerization were developed in this paper. The conversion efficacy of thiol–ene systems was studied for various propagation to chain transfer kinetic rate-ratio (RK), and thiol–ene concentration molar-ratio (RC). Numerical data were analyzed using analytical formulas and compared with the experimental data. We demonstrated that our model for a thiol–acrylate system with homopolymerization effects, and for a thiol–norbornene system with viscosity effects, fit much better with the measured data than a previous model excluding these effects. The general features for the roles of RK and RC on the conversion efficacy of thiol (CT) and ene (CV) are: (i) for RK = 1, CV and CT have the same temporal profiles, but have a reversed dependence on RC; (ii) for RK >> 1, CT are almost independent of RC; (iii) for RK << 1, CV and CT have the same profiles and both are decreasing functions of the homopolymerization effects defined by kCV; (iv) viscosity does not affect the efficacy in the case of RK >> 1, but reduces the efficacy of CV for other values of RK. -
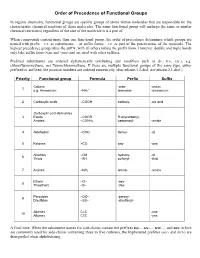
Priority of Functional Groups
Order of Precedence of Functional Groups In organic chemistry, functional groups are specific groups of atoms within molecules that are responsible for the characteristic chemical reactions of those molecules. The same functional group will undergo the same or similar chemical reaction(s) regardless of the size of the molecule it is a part of. When compounds contain more than one functional group, the order of precedence determines which groups are named with prefix – i.e. as substituents –, or suffix forms – i.e. as part of the parent name of the molecule. The highest precedence group takes the suffix, with all others taking the prefix form. However, double and triple bonds only take suffix form (-ene and -yne) and are used with other suffixes. Prefixed substituents are ordered alphabetically (excluding any modifiers such as di-, tri-, etc.), e.g. chlorofluoromethane, not fluorochloromethane. If there are multiple functional groups of the same type, either prefixed or suffixed, the position numbers are ordered numerically (thus ethane-1,2-diol, not ethane-2,1-diol.) Priority Functional group Formula Prefix Suffix Cations -onio- -onium 1 + e.g. Ammonium –NH4 ammonio- -ammonium 2 Carboxylic acids –COOH carboxy- -oic acid Carboxylic acid derivatives 3 Esters –COOR R-oxycarbonyl- Amides –CONH2 carbamoyl- -amide 4 Aldehydes –CHO formyl- -al 5 Ketones >CO oxo- -one Alcohols –OH hydroxy- -ol 6 Thiols –SH sulfanyl- -thiol 7 Amines –NH2 amino- -amine Ethers –O– -oxy- 8 Thioethers –S– -thio- Peroxides –OO– -peroxy- 9 Disulfides –SS– -disulfanyl- Alkenes C=C -ene 10 Alkynes C≡C -yne A final note: When the substituent names for side-chains contain the prefixes iso..., sec-..., tert-..., and neo (which are commonly used for side-chains containing three to five carbons), the hyphenated prefixes (sec- and tert-) are disregarded when alphabetizing. -

CHAPTER 7 ALCOHOLS, THIOLS, PHENOLS, ETHERS 7.1 Alcohols
CHAPTER 7 ALCOHOLS, THIOLS, PHENOLS, ETHERS Several new functional groups are presented in this chapter. All of the functions are based on oxygen and sulfur in the sp2 hybridized state. The functional groups contain two pairs of non-bonding electrons and are the cornerstone of many organic processes. The structures for alcohols, phenols, thiols, ethers and thioethers are shown below. Alkyl-OH Ph-OH Alkyl-SH R-O-R R-S-R alcohol phenol thiol ether thioether 7.1 Alcohols 7.1a Nomenclature Priorities in nomenclature Several functional groups have been encountered as we have advanced through the chapters. When a new functional group is presented, its nomenclature is always based on the parent name with an ending that designates the functional group. When several functional groups are present in a molecule, priority rules must be used to determine which function is the parent system that determines the numbering of the system and the suffix used in the final name. The priority list is as follows: carboxylic acids > aldehydes> ketones> alcohols > amines > alkene > alkyne > alkyl, halogen, nitro Thus a compound that contained hydroxy, alkene and ketone functions would be named as a ketone with the ketone getting the lowest number possible, and substituents are numbered accordingly and named in alphabetical order. Below the compound on the left is named as an alcohol, but the one on the right is named as a ketone even though both compounds have the seven carbon backbone. 124 Ch 7 Alcohols, Thiols, Phenols, Ethers OH OH Cl Cl O 7-chloro-3-hepten-2-ol 1-chloro-6-hydroxy-4-hepten-3-one OH has priority ketone has priority Alcohol Nomenclature Hydroxy compounds are encountered frequently in organic chemistry and the OH function is of high priority with only acids, aldehydes and ketones having higher priority. -

Thiol-Yne Click Polymerization
Review SPECIAL ISSUE: Molecular Materials and Devices August 2013 Vol.58 No.22: 27112718 doi: 10.1007/s11434-013-5892-1 Thiol-yne click polymerization YAO BiCheng1, SUN JingZhi1, QIN AnJun1* & TANG Ben Zhong1,2* 1 MOE Key Laboratory of Macromolecular Synthesis and Functionalization, Department of Polymer Science and Engineering, Zhejiang University, Hangzhou 310027, China; 2 Department of Chemistry, Institute for Advanced Study, Institute of Molecular Functional Materials, The Hong Kong University of Science & Technology, Clear Water Bay, Kowloon, Hong Kong, China Received February 1, 2013; accepted April 22, 2013; published online June 6, 2013 The research on using thiol-ene click reaction to synthesize sulfur-containing polymers with topological structures and advanced functional properties is a hot topic. However, the application of the thiol-yne reaction in the functional polymer preparation is limited and the thiol-yne click polymerization is to be further developed. In this review, we summarized recent research efforts on using thiol-yne click polymerization to synthesize polymers with topological structures. The sulfur-containing polymers were facilely prepared by photo- and thermo-initiated, amine-mediated, and transition-metal-catalyzed thiol-yne click polymerizations. These polymers are promising to be used as drug-delivery vehicles, high refractive index optical materials, photovoltaic materials, and biomaterials etc. thiol-yne click polymerization, radical mechanism, amine, rhodium complex, function Citation: Yao B C, Sun J Z, Qin A J, et al. Thiol-yne click polymerization. Chin Sci Bull, 2013, 58: 27112718, doi: 10.1007/s11434-013-5892-1 The exploration of new polymerization method for the group via well-known two-step processes, and fully additive preparation of functional polymers with unique structures is products with branched structures will be yielded [16].