Distinct Mutational Signatures Characterize Concurrent Loss of Polymerase Proofreading and Mismatch Repair
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Emergence of DNA Polymerase E Antimutators That Escape Error-Induced Extinction in Yeast
INVESTIGATION Emergence of DNA Polymerase e Antimutators That Escape Error-Induced Extinction in Yeast Lindsey N. Williams, Alan J. Herr, and Bradley D. Preston1 Department of Pathology, University of Washington, Seattle, Washington 98195 ABSTRACT DNA polymerases (Pols) e and d perform the bulk of yeast leading- and lagging-strand DNA synthesis. Both Pols possess intrinsic proofreading exonucleases that edit errors during polymerization. Rare errors that elude proofreading are extended into duplex DNA and excised by the mismatch repair (MMR) system. Strains that lack Pol proofreading or MMR exhibit a 10- to 100-fold increase in spontaneous mutation rate (mutator phenotype), and inactivation of both Pol d proofreading (pol3-01) and MMR is lethal due to replication error-induced extinction (EEX). It is unclear whether a similar synthetic lethal relationship exists between defects in Pol e proofreading (pol2-4) and MMR. Using a plasmid-shuffling strategy in haploid Saccharomyces cerevisiae, we observed synthetic lethality of pol2-4 with alleles that completely abrogate MMR (msh2D, mlh1D, msh3D msh6D,orpms1D mlh3D) but not with partial MMR loss (msh3D, msh6D, pms1D,ormlh3D), indicating that high levels of unrepaired Pol e errors drive extinction. However, variants that escape this error-induced extinction (eex mutants) frequently emerged. Five percent of pol2-4 msh2D eex mutants encoded second-site changes in Pol e that reduced the pol2-4 mutator phenotype between 3- and 23-fold. The remaining eex alleles were extragenic to pol2-4. The locations of antimutator amino-acid changes in Pol e and their effects on mutation spectra suggest multiple mechanisms of mutator suppression. Our data indicate that unrepaired leading- and lagging-strand polymerase errors drive extinction within a few cell divisions and suggest that there are polymerase-specific pathways of mutator suppression. -

Mutations That Separate the Functions of the Proofreading Subunit of the Escherichia Coli Replicase
G3: Genes|Genomes|Genetics Early Online, published on April 15, 2015 as doi:10.1534/g3.115.017285 Mutations that separate the functions of the proofreading subunit of the Escherichia coli replicase Zakiya Whatley*,1 and Kenneth N Kreuzer*§ *University Program in Genetics & Genomics, Duke University, Durham, NC 27705 §Department of Biochemistry, Duke University Medical Center, Durham, NC 27710 1 © The Author(s) 2013. Published by the Genetics Society of America. Running title: E. coli dnaQ separation of function mutants Keywords: DNA polymerase, epsilon subunit, linker‐scanning mutagenesis, mutation rate, SOS response Corresponding author: Kenneth N Kreuzer, Department of Biochemistry, Box 3711, Nanaline Duke Building, Research Drive, Duke University Medical Center, Durham, NC 27710 Phone: 919 684 6466 FAX: 919 684 6525 Email: [email protected] 1 Present address: Department of Biology, 300 N Washington Street, McCreary Hall, Campus Box 392, Gettysburg College, Gettysburg, PA 17325 Phone: 717 337 6160 Fax: 7171 337 6157 Email: [email protected] 2 ABSTRACT The dnaQ gene of Escherichia coli encodes the ε subunit of DNA polymerase III, which provides the 3’ 5’ exonuclease proofreading activity of the replicative polymerase. Prior studies have shown that loss of ε leads to high mutation frequency, partially constitutive SOS, and poor growth. In addition, a previous study from our lab identified dnaQ knockout mutants in a screen for mutants specifically defective in the SOS response following quinolone (nalidixic acid) treatment. To explain these results, we propose a model whereby in addition to proofreading, ε plays a distinct role in replisome disassembly and/or processing of stalled replication forks. -

The Architecture of a Eukaryotic Replisome
The Architecture of a Eukaryotic Replisome Jingchuan Sun1,2, Yi Shi3, Roxana E. Georgescu3,4, Zuanning Yuan1,2, Brian T. Chait3, Huilin Li*1,2, Michael E. O’Donnell*3,4 1 Biosciences Department, Brookhaven National Laboratory, Upton, New York, USA 2 Department of Biochemistry & Cell Biology, Stony Brook University, Stony Brook, New York, USA. 3 The Rockefeller University, 1230 York Avenue, New York, New York, USA. 4 Howard Hughes Medical Institute *Correspondence and requests for materials should be addressed to M.O.D. ([email protected]) or H.L. ([email protected]) ABSTRACT At the eukaryotic DNA replication fork, it is widely believed that the Cdc45-Mcm2-7-GINS (CMG) helicase leads the way in front to unwind DNA, and that DNA polymerases (Pol) trail behind the helicase. Here we use single particle electron microscopy to directly image a replisome. Contrary to expectations, the leading strand Pol ε is positioned ahead of CMG helicase, while Ctf4 and the lagging strand Pol α-primase (Pol α) are behind the helicase. This unexpected architecture indicates that the leading strand DNA travels a long distance before reaching Pol ε, it first threads through the Mcm2-7 ring, then makes a U-turn at the bottom to reach Pol ε at the top of CMG. Our work reveals an unexpected configuration of the eukaryotic replisome, suggests possible reasons for this architecture, and provides a basis for further structural and biochemical replisome studies. INTRODUCTION DNA is replicated by a multi-protein machinery referred to as a replisome 1,2. Replisomes contain a helicase to unwind DNA, DNA polymerases that synthesize the leading and lagging strands, and a primase that makes short primed sites to initiate DNA synthesis on both strands. -

Teacher Notes Science Nspired
DNA Replication TEACHER NOTES SCIENCE NSPIRED Science Objectives In this lesson, students will • Explore how the structure of DNA supports its semi-conservative replication • Identify the name and function of several key enzymes in DNA replication • Recognize the function of Okazaki fragments in DNA replication Vocabulary TI-Nspire™ Technology Skills: • Double helix • Leading strand • Download a TI-Nspire • Lagging strand • Polymerase I • Polymerase III • Helicase document • Base pairs • Okazaki Fragments • Open a document • Primase • Move between pages • Open Directions Box About the Lesson • Explore ‘Hot Spots’ • Using three simulations, students will interact with DNA replication to explore semi-conservative replication and identify Tech Tips: specific enzymes and their roles in replication. Assessments are Make sure that students know embedded in the activity to engage discussion and gauge how to move between pages by learning. pressing /¡ (left arrow) and • As a result, students will: /¢ (right arrow). • Learn the basic functions of the following DNA replication enzymes: helicase, primase, ligase, polymerase I and III. Lesson Materials: • Learn how the double helix reproduces in a semi- Student Activity • DNA_Replication_Student.do conservative fashion c • Learn the role and function of Okazaki fragments in • DNA_Replication_Student.pd replication of the lagging strand. f TI-Nspire document TI-Nspire™ Navigator™ • DNA_Replication.tns Not required. If using Navigator: • Send out the DNA_Replication.tns file. • Monitor student progress -

Functional Characterization of the DNA Polymerase Epsilon and Its Involvement in the Maintenance of Genome Integrity in Arabidopsis Jose Antonio Pedroza-Garcia
Functional characterization of the DNA Polymerase epsilon and its involvement in the maintenance of genome integrity in Arabidopsis Jose Antonio Pedroza-Garcia To cite this version: Jose Antonio Pedroza-Garcia. Functional characterization of the DNA Polymerase epsilon and its involvement in the maintenance of genome integrity in Arabidopsis. Plants genetics. Université Paris Saclay (COmUE), 2016. English. NNT : 2016SACLS248. tel-03092326 HAL Id: tel-03092326 https://tel.archives-ouvertes.fr/tel-03092326 Submitted on 2 Jan 2021 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. NNT : 2016SACLS248 THESE DE DOCTORAT DE L’UNIVERSITE PARIS-SACLAY, préparée à l’Université Paris-Sud ÉCOLE DOCTORALE N° 567 Sciences du Végétal : du Gène à l’Ecosystème Spécialité de doctorat: Biologie Par M. José Antonio Pedroza-Garcia Functional characterization of the DNA Polymerase epsilon and its involvement in the maintenance of genome integrity in Arabidopsis Thèse présentée et soutenue à Orsay, le 22 septembre 2016 : Composition du Jury : M. Frugier, Florian Directeur de Recherche, CNRS Président Mme Gallego, Maria Professeure, Université Blaise Pascal Rapporteur M. Bendahmane, Mohammed Directeur de Recherche, INRA Rapporteur Mme Mézard, Christine Directrice de Recherche, CNRS Examinatrice Mme Chabouté, Marie-Edith Directrice de Recherche, CNRS Examinatrice Mme Raynaud, Cécile Chargée de Recherche, CNRS Directrice de thèse Acknowledgments First, I would like to express my huge gratitude to my supervisor, Cécile Raynaud. -
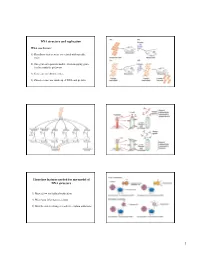
DNA Structure and Replication Three Key Features Needed for Any Model
DNA structure and replication What was known? 1) Hereditary factors were associated with specific traits 2) One-gene-one-protein model - from mapping genes for biosynthetic pathways 3) Genes are on chromosomes 4) Chromosomes are made up of DNA and protein Three key features needed for any model of DNA structure 1) Must allow for faithful replication 2) Must have information content 3) Must be able to change in order to explain mutations 1 Rosalind Franklin James Watson Francis Crick X-ray diffraction Maurice Wilkins Photograph of B-DNA Linus Pauling Nature 171, 737-738 (1953) © Macmillan Publishers Ltd. Molecular structure of Nucleic Acids WATSON, J. D. & CRICK, F. H. C. Features of the double helix A Structure for Deoxyribose Nucleic Acid 1) Two parallel strands We wish to suggest a structure for the salt of deoxyribose nucleic acid (D.N.A.). This structure has novel features which are of considerable biological interest………… 2) Bases held together by H-bonds 3) Phosphodiester backbone 4) The base is attached to the position 1 on the sugar 5) Base pair stack - provides Figure 1. This figure is purely diagrammatic. The two ribbons symbolize the two phophate-sugar chains, and the horizonal rods stability the pairs of bases holding the chains together. The vertical line marks the fibre axis. 6) Contains a major and …………….It has not escaped our notice that the specific pairing we minor groove have postulated immediately suggests a possible copying mechanism for the genetic material. Three key features needed for any model of DNA structure -

Processivity of DNA Polymerases: Two Mechanisms, One Goal Zvi Kelman1*, Jerard Hurwitz1 and Mike O’Donnell2
Minireview 121 Processivity of DNA polymerases: two mechanisms, one goal Zvi Kelman1*, Jerard Hurwitz1 and Mike O’Donnell2 Replicative DNA polymerases are highly processive Processive DNA synthesis by cellular replicases and the enzymes that polymerize thousands of nucleotides without bacteriophage T4 replicase dissociating from the DNA template. The recently Until recently, the only mechanism for high processivity determined structure of the Escherichia coli bacteriophage that was understood in detail was that utilized by cellular T7 DNA polymerase suggests a unique mechanism that replicases and the replicase of bacteriophage T4. This underlies processivity, and this mechanism may generalize mechanism involves a ring-shaped protein called a ‘DNA to other replicative polymerases. sliding clamp’ that encircles the DNA and tethers the polymerase catalytic unit to the DNA [3,4]. The three- Addresses: 1Department of Molecular Biology, Memorial Sloan- dimensional structures of several sliding clamps have been Kettering Cancer Center, 1275 York Avenue, New York, NY 10021, 2 determined: the eukaryotic proliferating cell nuclear USA and Laboratory of DNA Replication, Howard Hughes Medical β Institute, The Rockefeller University, 1230 York Avenue, New York, NY antigen (PCNA) [5,6]; the subunit of the prokaryotic 10021, USA. DNA polymerase III [7]; and the bacteriophage T4 gene 45 protein (gp45) (J Kuriyan, personal communication) *Corresponding author. (Figure 1). The overall structure of these clamps is very E-mail: [email protected] similar; the PCNA, β subunit and gp45 rings are super- Structure 15 February 1998, 6:121–125 imposable [8]. Each ring has similar dimensions and a http://biomednet.com/elecref/0969212600600121 central cavity large enough to accommodate duplex DNA (Figure 1). -

Primer Release Is the Rate-Limiting Event in Lagging-Strand Synthesis
Primer release is the rate-limiting event in lagging- strand synthesis mediated by the T7 replisome Alfredo J. Hernandeza, Seung-Joo Leea, and Charles C. Richardsona,1 aDepartment of Biological Chemistry and Molecular Pharmacology, Harvard Medical School, Boston, MA 02115 Contributed by Charles C. Richardson, April 18, 2016 (sent for review December 30, 2015; reviewed by Nicholas E. Dixon and I. Robert Lehman) DNA replication occurs semidiscontinuously due to the antiparallel but also for enabling the use of short oligoribonucleotides by T7 DNA strands and polarity of enzymatic DNA synthesis. Although DNA polymerase. Critically, the primase domain also fulfills two the leading strand is synthesized continuously, the lagging strand additional roles apart from primer synthesis: it prevents disso- is synthesized in small segments designated Okazaki fragments. ciation of the extremely short tetramer, stabilizing it with the Lagging-strand synthesis is a complex event requiring repeated template, and it secures it in the polymerase active site (10, 12). cycles of RNA primer synthesis, transfer to the lagging-strand Here we show that the rate-limiting step in initiation of Okazaki polymerase, and extension effected by cooperation between DNA fragments by the T7 replisome is primer release from the primase primase and the lagging-strand polymerase. We examined events domain of gp4. In the absence of gp2.5, an additional step, distinct controlling Okazaki fragment initiation using the bacteriophage from primer release, also limits primer extension. The presence of T7 replication system. Primer utilization by T7 DNA polymerase is gp2.5 promotes efficient primer formation and primer utilization. slower than primer formation. -

Junction Ribonuclease: an Activity in Okazaki Fragment Processing
Proc. Natl. Acad. Sci. USA Vol. 95, pp. 2244–2249, March 1998 Biochemistry Junction ribonuclease: An activity in Okazaki fragment processing RICHARD S. MURANTE,LEIGH A. HENRICKSEN, AND ROBERT A. BAMBARA* Department of Biochemistry and Biophysics, and Cancer Center, Box 712, University of Rochester School of Medicine and Dentistry, 601 Elmwood Avenue, Rochester, NY 14642 Communicated by I. Robert Lehman, Stanford University School of Medicine, Stanford, CA, December 29, 1997 (received for review September 15, 1997) ABSTRACT The initiator RNAs of mammalian Okazaki and removal of RNA primers. Reconstitution assays with both fragments are thought to be removed by RNase HI and the mouse cell and SV40 replication proteins require RNase HI for 5*-3* flap endonuclease (FEN1). Earlier evidence indicated the maturation of lagging strands in vitro (12, 13). that the cleavage site of RNase HI is 5* of the last ribonucle- Although cleavage of long RNAyDNA hybrids is random, otide at the RNA-DNA junction on an Okazaki substrate. In cleavage of certain substrates is clearly structure specific. Eder current work, highly purified calf RNase HI makes this exact et al. (16) found that when a series of ribonucleotides were cleavage in Okazaki fragments containing mismatches that followed by a 39 DNA segment and annealed to DNA to form distort the hybrid structure of the heteroduplex. Furthermore, a duplex, human RNase HI preferentially cleaved to leave a even fully unannealed Okazaki fragments were cleaved. DNA segment with a 59 monoribonucleotide. In reactions Clearly, the enzyme recognizes the transition from RNA to reconstituting Okazaki fragment processing, RNase HI from DNA on a single-stranded substrate and not the RNAyDNA calf thymus similarly cleaved the RNA-DNA strand 59 to the heteroduplex structure. -

The Possible Roles for Polyamines in the Initiation Process of SV40 DNA Replication in Vitro
535-539 9/1/08 14:46 Page 535 ONCOLOGY REPORTS 19: 535-539, 2008 535 The possible roles for polyamines in the initiation process of SV40 DNA replication in vitro DONG-GIL KIM1, JUAN DU1, CHUNHUI MIAO1, JEE H. JUNG3, SANG CHUL PARK4 and DONG-KYOO KIM1,2 1Department of Biomedicinal Chemistry, and Institute of Functional Materials, 2Biohealth Product Research Center, Inje University, Kimhae 621-749; 3College of Pharmacy, Pusan National University, Busan 609-735; 4Department of Biochemistry and Molecular Biology, The Aging and Apoptosis Research Center, Seoul National University College of Medicine, Seoul 110-799, Korea Received May 14, 2007; Accepted July 25, 2007 Abstract. The polyamines are aliphatic cations which are polyanionic macromolecules such as DNA (3). The most present in millimolar concentrations in all mammalian cells, obvious specific characteristic of the polyamines is their and are required for optimal growth of almost all cell types. polybasic character which gives them a much higher affinity In this study, the roles of polyamines in DNA replication for acidic constituents than that exhibited by Na+, K+, in vitro and the mechanism by which polyamines affected Mg2+, Ca2+, or monoamines; this polybasic character is most DNA replication were examined using simian virus 40 DNA pronounced with spermine because of its four positive groups. replication system in vitro. We found that polyamines The polyamines are required for optimal growth of almost all inhibited DNA replication, but it is not clear at which stage cell types and increased biosynthesis is necessary for the this occurs. Spermidine inhibited the DNA cleavage by traverse of a cell through the cell cycle. -

DNA Polymerases at the Eukaryotic Replication Fork Thirty Years After: Connection to Cancer
cancers Review DNA Polymerases at the Eukaryotic Replication Fork Thirty Years after: Connection to Cancer Youri I. Pavlov 1,2,* , Anna S. Zhuk 3 and Elena I. Stepchenkova 2,4 1 Eppley Institute for Research in Cancer and Allied Diseases and Buffett Cancer Center, University of Nebraska Medical Center, Omaha, NE 68198, USA 2 Department of Genetics and Biotechnology, Saint-Petersburg State University, 199034 Saint Petersburg, Russia; [email protected] 3 International Laboratory of Computer Technologies, ITMO University, 197101 Saint Petersburg, Russia; [email protected] 4 Laboratory of Mutagenesis and Genetic Toxicology, Vavilov Institute of General Genetics, Saint-Petersburg Branch, Russian Academy of Sciences, 199034 Saint Petersburg, Russia * Correspondence: [email protected] Received: 30 September 2020; Accepted: 13 November 2020; Published: 24 November 2020 Simple Summary: The etiology of cancer is linked to the occurrence of mutations during the reduplication of genetic material. Mutations leading to low replication fidelity are the culprits of many hereditary and sporadic cancers. The archetype of the current model of replication fork was proposed 30 years ago. In the sequel to our 2010 review with the words “years after” in the title inspired by A. Dumas’s novels, we go over new developments in the DNA replication field and analyze how they help elucidate the effects of the genetic variants of DNA polymerases on cancer. Abstract: Recent studies on tumor genomes revealed that mutations in genes of replicative DNA polymerases cause a predisposition for cancer by increasing genome instability. The past 10 years have uncovered exciting details about the structure and function of replicative DNA polymerases and the replication fork organization. -
![The Second Subunit of DNA Polymerase Delta Is Required for Genomic Stability and Epigenetic Regulation1[OPEN]](https://docslib.b-cdn.net/cover/7861/the-second-subunit-of-dna-polymerase-delta-is-required-for-genomic-stability-and-epigenetic-regulation1-open-1617861.webp)
The Second Subunit of DNA Polymerase Delta Is Required for Genomic Stability and Epigenetic Regulation1[OPEN]
The Second Subunit of DNA Polymerase Delta Is Required for Genomic Stability and Epigenetic Regulation1[OPEN] Jixiang Zhang, Shaojun Xie, Jinkui Cheng, Jinsheng Lai, Jian-Kang Zhu, and Zhizhong Gong* State Key Laboratory of Plant Physiology and Biochemistry, College of Biological Sciences, China Agricultural University, Beijing 100193, China (J.Z., J.C., Z.G.); Shanghai Center for Plant Stress Biology, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, Shanghai 200032, China (S.X., J.-K.Z.); Department of Horticulture and Landscape Architecture, Purdue University, West Lafayette, Indiana 47906 (S.X., J.-K.Z.); and State Key Laboratory of Agrobiotechnology, China National Maize Improvement Center, Department of Plant Genetics and Breeding, China Agricultural University, Beijing 100193, China (J.L.) ORCID IDs: 0000-0002-1641-8650 (J.Z.); 0000-0002-6719-9814 (S.X.); 0000-0001-5134-731X (J.-K.Z.). DNA polymerase d plays crucial roles in DNA repair and replication as well as maintaining genomic stability. However, the function of POLD2, the second small subunit of DNA polymerase d, has not been characterized yet in Arabidopsis (Arabidopsis thaliana). During a genetic screen for release of transcriptional gene silencing, we identified a mutation in POLD2. Whole-genome bisulfite sequencing indicated that POLD2 is not involved in the regulation of DNA methylation. POLD2 genetically interacts with Ataxia Telangiectasia-mutated and Rad3-related and DNA polymerase a. The pold2-1 mutant exhibits genomic instability with a high frequency of homologous recombination. It also exhibits hypersensitivity to DNA-damaging reagents and short telomere length. Whole-genome chromatin immunoprecipitation sequencing and RNA sequencing analyses suggest that pold2-1 changes H3K27me3 and H3K4me3 modifications, and these changes are correlated with the gene expression levels.