Mouse Models of Syndromic Craniosynostosis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Increased Nuchal Translucency Precision Panel
Increased Nuchal Translucency Precision Panel Overview Increased Nuchal Translucency (NT) is defined as an abnormal accumulation of fluid in the nuchal area, which is visualized as a thickened sonolucent area. It is a standardized measure obtained between 11 and 14 weeks of gestation to calculate the risk of a fetus being affected by a chromosomal aneuploidy. NT>3.5mm has been found to be associated with fetal chromosomal abnormalities and single-gene disorders as well as cardiac defects and other structural abnormalities in euploid and aneuploid fetuses. Proportionally as NT increases, even with a normal karyotype, there is a higher risk of adverse pregnancy outcomes such as miscarriage, intrauterine death, congenital heart defects and numerous other structural and genetic syndromes. There is not one single cause of increased NT, it is based on a complex and multifactorial process, linked to one or more embryonic processes. It has been shown that a persistently increased NT with a normal karyotype and aCGH has a 4-10% probability of being associated to Noonan Syndrome and/or other RASopathies using Whole Exome Sequencing (WES). However, the general tendency following detection of isolated enlarged NT in an euploid fetus is that most babies with normal detailed ultrasound examination and echocardiography will have uneventful outcomes. The Igenomix Increased Nuchal Translucency Precision Panel can be used to make a directed and accurate prenatal differential diagnosis of increased nuchal translucency in patients with or without a normal karyotype ultimately leading to a better management and prognosis of the associated comorbidities. It provides a comprehensive analysis of the genes involved in this disease using next-generation sequencing (NGS) to fully understand the spectrum of relevant genes involved. -
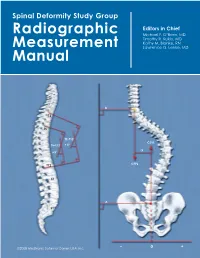
Spinal Deformity Study Group
Spinal Deformity Study Group Editors in Chief Radiographic Michael F. O’Brien, MD Timothy R. Kuklo, MD Kathy M. Blanke, RN Measurement Lawrence G. Lenke, MD Manual B T2 T5 T2–T12 CSVL T5–T12 +X° -X +X° C7PL T12 L2 A S1 ©2008 Medtronic Sofamor Danek USA, Inc. – 0 + Radiographic Measurement Manual Editors in Chief Michael F. O’Brien, MD Timothy R. Kuklo, MD Kathy M. Blanke, RN Lawrence G. Lenke, MD Section Editors Keith H. Bridwell, MD Kathy M. Blanke, RN Christopher L. Hamill, MD William C. Horton, MD Timothy R. Kuklo, MD Hubert B. Labelle, MD Lawrence G. Lenke, MD Michael F. O’Brien, MD David W. Polly Jr, MD B. Stephens Richards III, MD Pierre Roussouly, MD James O. Sanders, MD ©2008 Medtronic Sofamor Danek USA, Inc. Acknowledgements Radiographic Measurement Manual The radiographic measurement manual has been developed to present standardized techniques for radiographic measurement. In addition, this manual will serve as a complimentary guide for the Spinal Deformity Study Group’s radiographic measurement software. Special thanks to the following members of the Spinal Deformity Study Group in the development of this manual. Sigurd Berven, MD Hubert B. Labelle, MD Randal Betz, MD Lawrence G. Lenke, MD Fabien D. Bitan, MD Thomas G. Lowe, MD John T. Braun, MD John P. Lubicky, MD Keith H. Bridwell, MD Steven M. Mardjetko, MD Courtney W. Brown, MD Richard E. McCarthy, MD Daniel H. Chopin, MD Andrew A. Merola, MD Edgar G. Dawson, MD Michael Neuwirth, MD Christopher DeWald, MD Peter O. Newton, MD Mohammad Diab, MD Michael F. -

Lordosis, Kyphosis, and Scoliosis
SPINAL CURVATURES: LORDOSIS, KYPHOSIS, AND SCOLIOSIS The human spine normally curves to aid in stability or balance and to assist in absorbing shock during movement. These gentle curves can be seen from the side or lateral view of the spine. When viewed from the back, the spine should run straight down the middle of the back. When there are abnormalities or changes in the natural spinal curvature, these abnormalities are named with the following conditions and include the following symptoms. LORDOSIS Some lordosis is normal in the lower portion or, lumbar section, of the human spine. A decreased or exaggerated amount of lordosis that is causing spinal instability is a condition that may affect some patients. Symptoms of Lordosis include: ● Appearance of sway back where the lower back region has a pronounced curve and looks hollow with a pronounced buttock area ● Difficulty with movement in certain directions ● Low back pain KYPHOSIS This condition is diagnosed when the patient has a rounded upper back and the spine is bent over or curved more than 50 degrees. Symptoms of Kyphosis include: ● Curved or hunched upper back ● Patient’s head that leans forward ● May have upper back pain ● Experiences upper back discomfort after movement or exercise SCOLIOSIS The most common of the three curvatures. This condition is diagnosed when the spine looks like a “s” or “c” from the back. The spine is not straight up and down but has a curve or two running side-to-side. Sagittal Balance Definition • Sagittal= front-to-back direction (sagittal plane) • Imbalance= Lack of harmony or balance Etiology • Excessive lordosis (backwards lean) or kyphosis (forward lean) • Traumatic injury • Previous spinal fusion that disrupted sagittal balance Effects • Low back pain • Difficulty walking • Inability to look straight ahead when upright The most ergonomic and natural posture is to maintain neutral balance, with the head positioned over the shoulders and pelvis. -

Saethre-Chotzen Syndrome
Saethre-Chotzen syndrome Authors: Professor L. Clauser1 and Doctor M. Galié Creation Date: June 2002 Update: July 2004 Scientific Editor: Professor Raoul CM. Hennekam 1Department of craniomaxillofacial surgery, St. Anna Hospital and University, Corso Giovecca, 203, 44100 Ferrara, Italy. [email protected] Abstract Keywords Disease name and synonyms Excluded diseases Definition Prevalence Management including treatment Etiology Diagnostic methods Genetic counseling Antenatal diagnosis Unresolved questions References Abstract Saethre-Chotzen Syndrome (SCS) is an inherited craniosynostotic condition, with both premature fusion of cranial sutures (craniostenosis) and limb abnormalities. The most common clinical features, present in more than a third of patients, consist of coronal synostosis, brachycephaly, low frontal hairline, facial asymmetry, hypertelorism, broad halluces, and clinodactyly. The estimated birth incidence is 1/25,000 to 1/50,000 but because the phenotype can be very mild, the entity is likely to be underdiagnosed. SCS is inherited as an autosomal dominant trait with a high penetrance and variable expression. The TWIST gene located at chromosome 7p21-p22, is responsible for SCS and encodes a transcription factor regulating head mesenchyme cell development during cranial tube formation. Some patients with an overlapping SCS phenotype have mutations in the FGFR3 (fibroblast growth factor receptor 3) gene; especially the Pro250Arg mutation in FGFR3 (Muenke syndrome) can resemble SCS to a great extent. Significant intrafamilial -

MEDICAL GENETICS RESIDENCY PROGRAM Department of Pediatrics
MEDICAL GENETICS RESIDENCY PROGRAM Department of Pediatrics University of Michigan Health Systems (734) 763-6767 1500 E. Medical Center Drive (734) 763-6561 (fax) D5240 MPB Ann Arbor, MI 48109-5718 Biochemical Genetics Goals and Objectives Director: Drs. Ayesha Ahmad and Shane C. Quinonez The goals and objectives of the Biochemical Genetics Clinic rotation in the Medical Genetics Residency Program are to provide the resident with exposure to all aspects of care of metabolic disease and counseling in accordance with the Residency Review Committee for Medical Genetics expectations and to fulfill criteria for board eligibility by the American Board of Medical Genetics. Patient Care The resident will become familiar with the evaluation, diagnosis and management of urea cycle disorders, organic acidemias, disorders of carbohydrate and lipid metabolism and numerous other inborn errors of metabolism (IEMs). Residents will gain exposure in performing and expertise in interpreting biochemical analyses relevant to the diagnosis and management of human genetic diseases. By the end of the rotation the resident should be able to identify signs and symptoms of IEMs, formulate a differential diagnosis, order appropriate tests and recognize normal variants and complex patterns of metabolites. Residents will be able to manage acute metabolic crises and provide chronic management of patients with an IEM. Residents should be able to interpret NBS results, collaborate with the primary provider to act upon results in a timely manner, develop a differential diagnosis and order appropriate confirmatory testing and communicate results to families. Medical Knowledge Through coursework and didactic sessions with attending physicians, residents will become familiar with fundamental concepts, molecular biology and biochemistry relevant to IEMs. -

Kyphectomy in Neonates with Meningomyelocele
Child's Nervous System (2019) 35:673–681 https://doi.org/10.1007/s00381-018-4006-4 ORIGINAL PAPER Kyphectomy in neonates with meningomyelocele Nail Özdemir1 & Senem Alkan Özdemir2 & Esra Arun Özer3 Received: 18 August 2018 /Accepted: 18 November 2018 /Published online: 11 December 2018 # Springer-Verlag GmbH Germany, part of Springer Nature 2018 Abstract Purpose Kyphosis is the most severe spinal deformity associated with meningomyelocele (MMC) and is seen in approximately 15% of neonates. Our purpose is to present our clinical experience, to discuss the technique and deformity correction in kyphectomy in neonates with MMC, and to assess its long-term outcomes. Method In this prospective study, the authors reviewed eight cases submitted to surgery between 2013 and 2015. We evaluated clinical characteristics that were analyzed, as were the operative technique employed, and angle range of the kyphosis deformity postcorrection follow-up. Results Neonatal kyphectomy was performed of six females and two males. The mean birth weight was 2780 g, and the mean age at the time of surgery was 5.6 days. There were S-shaped type deformity in lumbar region in all neonates. In the correction of the kyphotic deformity, a total vertebrae were removed from four patient, whereas a partial vertebrectomy was done in four. The mean operative time was 116 min. No patients did not require the blood transfusion. There were no serious complications, and wound closure was successful in all patients. The mean follow-up period was 4 years and 3 months (range 36–61 months), except one patient who died 1 week after discharge. -

Scoliosis in Paraplegia
Paraplegia (1974), II, 290-292 SCOLIOSIS IN PARAPLEGIA By JOHN A. ODOM, JR., M.D. and COURTN EY W. BROWN, M.D. Children's Hospital, Denver, Colorado in conjunction with ROBERT R. JACKSON, M.D., HARRY R. HA HN, M.D. and TERRY V. CARLE, M.D. Craig Rehabilitation Hospital, Englewood, Colorado WITH the increasing instance of excellent medical care, more children with traumatic paraplegia and myelomeningocele with paraplegia live to adulthood. In these two groups of patients there is a high instance of scoliosis, kyphosis and lordosis. Much attention in the past years has been placed on hips and feet but only in the last decade has there been much attention concentrated on the treatment of the spines of these patients. Most of this attention has been toward the patient with a traumatic paraplegia with development of scoliosis. Some attempts have been made at fusing the scoliotic spine of myelomeningo celes by Harrington Instrumentation but with a rather high instance of complica tions and failures. With the event of anterior instrumentation by Dr. Alan Dwyer of Sydney, Australia, the anterior approach to the spine is becoming widely accepted and very successfully used. This is especially valuable in the correction and fusion of the spine with no posterior elements from birth. MATERIAL FOR STUDY Between March of 1971 and June of 1973, there have been 26 paraplegics with scoliosis who have been cared for by the authors. All of these patients have either been myelomeningoceles with paraplegia or spinal cord injuries, all of whom have had scoliosis, kyphosis, or severe lordosis. -

Prevalence and Incidence of Rare Diseases: Bibliographic Data
Number 1 | January 2019 Prevalence and incidence of rare diseases: Bibliographic data Prevalence, incidence or number of published cases listed by diseases (in alphabetical order) www.orpha.net www.orphadata.org If a range of national data is available, the average is Methodology calculated to estimate the worldwide or European prevalence or incidence. When a range of data sources is available, the most Orphanet carries out a systematic survey of literature in recent data source that meets a certain number of quality order to estimate the prevalence and incidence of rare criteria is favoured (registries, meta-analyses, diseases. This study aims to collect new data regarding population-based studies, large cohorts studies). point prevalence, birth prevalence and incidence, and to update already published data according to new For congenital diseases, the prevalence is estimated, so scientific studies or other available data. that: Prevalence = birth prevalence x (patient life This data is presented in the following reports published expectancy/general population life expectancy). biannually: When only incidence data is documented, the prevalence is estimated when possible, so that : • Prevalence, incidence or number of published cases listed by diseases (in alphabetical order); Prevalence = incidence x disease mean duration. • Diseases listed by decreasing prevalence, incidence When neither prevalence nor incidence data is available, or number of published cases; which is the case for very rare diseases, the number of cases or families documented in the medical literature is Data collection provided. A number of different sources are used : Limitations of the study • Registries (RARECARE, EUROCAT, etc) ; The prevalence and incidence data presented in this report are only estimations and cannot be considered to • National/international health institutes and agencies be absolutely correct. -

Blueprint Genetics Craniosynostosis Panel
Craniosynostosis Panel Test code: MA2901 Is a 38 gene panel that includes assessment of non-coding variants. Is ideal for patients with craniosynostosis. About Craniosynostosis Craniosynostosis is defined as the premature fusion of one or more cranial sutures leading to secondary distortion of skull shape. It may result from a primary defect of ossification (primary craniosynostosis) or, more commonly, from a failure of brain growth (secondary craniosynostosis). Premature closure of the sutures (fibrous joints) causes the pressure inside of the head to increase and the skull or facial bones to change from a normal, symmetrical appearance resulting in skull deformities with a variable presentation. Craniosynostosis may occur in an isolated setting or as part of a syndrome with a variety of inheritance patterns and reccurrence risks. Craniosynostosis occurs in 1/2,200 live births. Availability 4 weeks Gene Set Description Genes in the Craniosynostosis Panel and their clinical significance Gene Associated phenotypes Inheritance ClinVar HGMD ALPL Odontohypophosphatasia, Hypophosphatasia perinatal lethal, AD/AR 78 291 infantile, juvenile and adult forms ALX3 Frontonasal dysplasia type 1 AR 8 8 ALX4 Frontonasal dysplasia type 2, Parietal foramina AD/AR 15 24 BMP4 Microphthalmia, syndromic, Orofacial cleft AD 8 39 CDC45 Meier-Gorlin syndrome 7 AR 10 19 EDNRB Hirschsprung disease, ABCD syndrome, Waardenburg syndrome AD/AR 12 66 EFNB1 Craniofrontonasal dysplasia XL 28 116 ERF Craniosynostosis 4 AD 17 16 ESCO2 SC phocomelia syndrome, Roberts syndrome -

Medical Genetics and Genomic Medicine in the United States of America
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by George Washington University: Health Sciences Research Commons (HSRC) Himmelfarb Health Sciences Library, The George Washington University Health Sciences Research Commons Pediatrics Faculty Publications Pediatrics 7-1-2017 Medical genetics and genomic medicine in the United States of America. Part 1: history, demographics, legislation, and burden of disease. Carlos R Ferreira George Washington University Debra S Regier George Washington University Donald W Hadley P Suzanne Hart Maximilian Muenke Follow this and additional works at: https://hsrc.himmelfarb.gwu.edu/smhs_peds_facpubs Part of the Genetics and Genomics Commons APA Citation Ferreira, C., Regier, D., Hadley, D., Hart, P., & Muenke, M. (2017). Medical genetics and genomic medicine in the United States of America. Part 1: history, demographics, legislation, and burden of disease.. Molecular Genetics and Genomic Medicine, 5 (4). http://dx.doi.org/10.1002/mgg3.318 This Journal Article is brought to you for free and open access by the Pediatrics at Health Sciences Research Commons. It has been accepted for inclusion in Pediatrics Faculty Publications by an authorized administrator of Health Sciences Research Commons. For more information, please contact [email protected]. GENETICS AND GENOMIC MEDICINE AROUND THE WORLD Medical genetics and genomic medicine in the United States of America. Part 1: history, demographics, legislation, and burden of disease Carlos R. Ferreira1,2 , Debra S. Regier2, Donald W. Hadley1, P. Suzanne Hart1 & Maximilian Muenke1 1National Human Genome Research Institute, National Institutes of Health, Bethesda, Maryland 2Rare Disease Institute, Children’s National Health System, Washington, District of Columbia Correspondence Carlos R. -

Psykisk Utviklingshemming Og Forsinket Utvikling
Psykisk utviklingshemming og forsinket utvikling Genpanel, versjon v03 Tabellen er sortert på gennavn (HGNC gensymbol) Navn på gen er iht. HGNC >x10 Andel av genet som har blitt lest med tilfredstillende kvalitet flere enn 10 ganger under sekvensering x10 er forventet dekning; faktisk dekning vil variere. Gen Gen (HGNC Transkript >10x Fenotype (symbol) ID) AAAS 13666 NM_015665.5 100% Achalasia-addisonianism-alacrimia syndrome OMIM AARS 20 NM_001605.2 100% Charcot-Marie-Tooth disease, axonal, type 2N OMIM Epileptic encephalopathy, early infantile, 29 OMIM AASS 17366 NM_005763.3 100% Hyperlysinemia OMIM Saccharopinuria OMIM ABCB11 42 NM_003742.2 100% Cholestasis, benign recurrent intrahepatic, 2 OMIM Cholestasis, progressive familial intrahepatic 2 OMIM ABCB7 48 NM_004299.5 100% Anemia, sideroblastic, with ataxia OMIM ABCC6 57 NM_001171.5 93% Arterial calcification, generalized, of infancy, 2 OMIM Pseudoxanthoma elasticum OMIM Pseudoxanthoma elasticum, forme fruste OMIM ABCC9 60 NM_005691.3 100% Hypertrichotic osteochondrodysplasia OMIM ABCD1 61 NM_000033.3 77% Adrenoleukodystrophy OMIM Adrenomyeloneuropathy, adult OMIM ABCD4 68 NM_005050.3 100% Methylmalonic aciduria and homocystinuria, cblJ type OMIM ABHD5 21396 NM_016006.4 100% Chanarin-Dorfman syndrome OMIM ACAD9 21497 NM_014049.4 99% Mitochondrial complex I deficiency due to ACAD9 deficiency OMIM ACADM 89 NM_000016.5 100% Acyl-CoA dehydrogenase, medium chain, deficiency of OMIM ACADS 90 NM_000017.3 100% Acyl-CoA dehydrogenase, short-chain, deficiency of OMIM ACADVL 92 NM_000018.3 100% VLCAD -

Differential Diagnosis Functional Vs Structural Scoliosis
Differential Diagnosis Functional vs Structural Scoliosis James J. Lehman, DC, MBA, FACO Associate Professor of Clinical Sciences University of Bridgeport College of Chiropractic Director Community Health Clinical Education University of Bridgeport Diagnosis is the key to successful treatment Scoliosis Classification Based upon the findings with this postural presentation, what physical examination procedures would you perform to determine your working diagnosis for the child with scoliosis? Classification of Scoliosis Structural or Nonstructural (functional) 1. Structural curves are fixed, nonflexible, and fail to correct with bending. 2. Nonstructural curves are not fixed but flexible and readily correct with bending. Postural Evaluation of Spine • Observation of standing posture • Right thoracic curve is most common with best prognosis Adam’s Position Differential Diagnosis Functional Scoliosis/Postural Imbalance Pelvic Obliquity and Postural Imbalance • You must determine whether the leg length discrepancy is anatomical or functional Actual Leg-Length Test • This is a tape measurement that tests for anatomical leg length discrepancy. • ASIS and medial malleolus are the landmarks identified Apparent Leg-Length Test • Reveals functional leg length discrepancy • Umbillicus and medial malleolus are landmarks • Evans Functional Leg-Length Measurement • Measure length of both lower extremities supine and seated • Inferior medial malloli are used as landmarks • Read the body language Functional Leg-Length Measurement • Usually the ipsilateral malleolus will measure short when supine if the superior iliac crest appears inferior when standing and long when seated Clinical Value of Long Sit Test • Pelvic Obliquity • Leg Length Discrepancy – Functional – Anatomical • SIJ Dysfunction • Spinal manipulation VIDEO: SUPINE TO LONG SIT TEST HTTP://WWW.THESTUDENTPHYSICALTHERAPIST.COM/SUPINE-TO-LONG- SIT-TEST.HTML LEVANGIE PK.