Identification of Novel Betaherpesviruses in Iberian Bats Reveals Parallel Evolution
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Special Publications Museum of Texas Tech University Number 63 18 September 2014
Special Publications Museum of Texas Tech University Number 63 18 September 2014 List of Recent Land Mammals of Mexico, 2014 José Ramírez-Pulido, Noé González-Ruiz, Alfred L. Gardner, and Joaquín Arroyo-Cabrales.0 Front cover: Image of the cover of Nova Plantarvm, Animalivm et Mineralivm Mexicanorvm Historia, by Francisci Hernández et al. (1651), which included the first list of the mammals found in Mexico. Cover image courtesy of the John Carter Brown Library at Brown University. SPECIAL PUBLICATIONS Museum of Texas Tech University Number 63 List of Recent Land Mammals of Mexico, 2014 JOSÉ RAMÍREZ-PULIDO, NOÉ GONZÁLEZ-RUIZ, ALFRED L. GARDNER, AND JOAQUÍN ARROYO-CABRALES Layout and Design: Lisa Bradley Cover Design: Image courtesy of the John Carter Brown Library at Brown University Production Editor: Lisa Bradley Copyright 2014, Museum of Texas Tech University This publication is available free of charge in PDF format from the website of the Natural Sciences Research Laboratory, Museum of Texas Tech University (nsrl.ttu.edu). The authors and the Museum of Texas Tech University hereby grant permission to interested parties to download or print this publication for personal or educational (not for profit) use. Re-publication of any part of this paper in other works is not permitted without prior written permission of the Museum of Texas Tech University. This book was set in Times New Roman and printed on acid-free paper that meets the guidelines for per- manence and durability of the Committee on Production Guidelines for Book Longevity of the Council on Library Resources. Printed: 18 September 2014 Library of Congress Cataloging-in-Publication Data Special Publications of the Museum of Texas Tech University, Number 63 Series Editor: Robert J. -

A Recent Bat Survey Reveals Bukit Barisan Selatan Landscape As A
A Recent Bat Survey Reveals Bukit Barisan Selatan Landscape as a Chiropteran Diversity Hotspot in Sumatra Author(s): Joe Chun-Chia Huang, Elly Lestari Jazdzyk, Meyner Nusalawo, Ibnu Maryanto, Maharadatunkamsi, Sigit Wiantoro, and Tigga Kingston Source: Acta Chiropterologica, 16(2):413-449. Published By: Museum and Institute of Zoology, Polish Academy of Sciences DOI: http://dx.doi.org/10.3161/150811014X687369 URL: http://www.bioone.org/doi/full/10.3161/150811014X687369 BioOne (www.bioone.org) is a nonprofit, online aggregation of core research in the biological, ecological, and environmental sciences. BioOne provides a sustainable online platform for over 170 journals and books published by nonprofit societies, associations, museums, institutions, and presses. Your use of this PDF, the BioOne Web site, and all posted and associated content indicates your acceptance of BioOne’s Terms of Use, available at www.bioone.org/page/terms_of_use. Usage of BioOne content is strictly limited to personal, educational, and non-commercial use. Commercial inquiries or rights and permissions requests should be directed to the individual publisher as copyright holder. BioOne sees sustainable scholarly publishing as an inherently collaborative enterprise connecting authors, nonprofit publishers, academic institutions, research libraries, and research funders in the common goal of maximizing access to critical research. Acta Chiropterologica, 16(2): 413–449, 2014 PL ISSN 1508-1109 © Museum and Institute of Zoology PAS doi: 10.3161/150811014X687369 A recent -

Chiroptera: Vespertilionidae) from Taiwan and Adjacent China
Zootaxa 3920 (1): 301–342 ISSN 1175-5326 (print edition) www.mapress.com/zootaxa/ Article ZOOTAXA Copyright © 2015 Magnolia Press ISSN 1175-5334 (online edition) http://dx.doi.org/10.11646/zootaxa.3920.2.6 http://zoobank.org/urn:lsid:zoobank.org:pub:8B991675-0C48-40D4-87D2-DACA524D17C2 Molecular phylogeny and morphological revision of Myotis bats (Chiroptera: Vespertilionidae) from Taiwan and adjacent China MANUEL RUEDI1,5, GÁBOR CSORBA2, LIANG- KONG LIN3 & CHENG-HAN CHOU3,4 1Department of Mammalogy and Ornithology, Natural History Museum of Geneva, Route de Malagnou 1, BP 6434, 1211 Geneva (6), Switzerland. E-mail: [email protected] 2Department of Zoology, Hungarian Natural History Museum, Budapest, Baross u. 13., H-1088. E-mail: [email protected] 3Laboratory of Wildlife Ecology, Department of Biology, Tunghai University, Taichung, Taiwan 407, R.O.C. E-mail: [email protected] 4Division of Zoology, Endemic Species Research Institute, Nantou, Taiwan 552, R.O.C. E-mail: [email protected] 5Corresponding author Table of contents Abstract . 301 Introduction . 302 Material and methods . 310 Results . 314 Discussion . 319 Systematic account . 319 Submyotodon latirostris (Kishida, 1932) . 319 Myotis fimbriatus (Peters, 1870) . 321 Myotis laniger (Peters, 1870) . 322 Myotis secundus sp. n. 324 Myotis soror sp. n. 327 Myotis frater Allen, 1923 . 331 Myotis formosus (Hodgson, 1835) . 334 Myotis rufoniger (Tomes, 1858) . 335 Biogeography and conclusions . 336 Key to the Myotinae from Taiwan and adjacent mainland China . 337 Acknowledgments . 337 References . 338 Abstract In taxonomic accounts, three species of Myotis have been traditionally reported to occur on the island of Taiwan: Watase’s bat (M. -

EU Action Plan for the Conservation of All Bat Species in the European Union
Action Plan for the Conservation of All Bat Species in the European Union 2018 – 2024 October 2018 Action Plan for the Conservation of All Bat Species in the European Union 2018 - 2024 EDITORS: BAROVA Sylvia (European Commission) & STREIT Andreas (UNEP/EUROBATS) COMPILERS: MARCHAIS Guillaume & THAURONT Marc (Ecosphère, France/The N2K Group) CONTRIBUTORS (in alphabetical order): BOYAN Petrov * (Bat Research & Conservation Centre, Bulgaria) DEKKER Jasja (Animal ecologist, Netherlands) ECOSPHERE: JUNG Lise, LOUTFI Emilie, NUNINGER Lise & ROUÉ Sébastien GAZARYAN Suren (EUROBATS) HAMIDOVIĆ Daniela (State Institute for Nature Protection, Croatia) JUSTE Javier (Spanish association for the study and conservation of bats, Spain) KADLEČÍK Ján (Štátna ochrana prírody Slovenskej republiky, Slovakia) KYHERÖINEN Eeva-Maria (Finnish Museum of Natural History, Finland) HANMER Julia (Bat Conservation Trust, United Kingdom) LEIVITS Meelis (Environmental Agency of the Ministry of Environment, Estonia) MARNELl Ferdia (National Parks & Wildlife Service, Ireland) PETERMANN Ruth (Federal Agency for Nature Conservation, Germany) PETERSONS Gunărs (Latvia University of Agriculture, Latvia) PRESETNIK Primož (Centre for Cartography of Fauna and Flora, Slovenia) RAINHO Ana (Institute for the Nature and Forest Conservation, Portugal) REITER Guido (Foundation for the protection of our bats in Switzerland) RODRIGUES Luisa (Institute for the Nature and Forest Conservation, Portugal) RUSSO Danilo (University of Napoli Frederico II, Italy) SCHEMBRI -

2010-7-Alcathoe's
An awful lot of bat workers are going to be looking even Alcathoe Bat more closely at any Myotis they encounter. You had to be there Top photo (c) http://www.krzysztof.piksa-pl.com As we arrived at the ZSL symposium people were in little Lower photo Derek Smith’s mystery bat huddles muttering excitedly. Soon we were as excited Prof John Altringham was using the symposium to announce that Britain has a new bat species The news was kept quiet until the results had been checked and double checked but there can be no doubt. We have discussed before the nightmare which is distinguishing Brandt and whiskered bats. One of John’s research students was doing genetic analysis of Brandt and Whiskered bats and contacted John because she was getting a sizeable cluster of anomalous results that fitted neither species when she looked at the gene sequences. DNA was taken from bats in the North York Moors and Susses, and when they also looked at samples from Europe, they got the same results John says that key features to look for are forearm length, and echolocation calls. Bob and Jude were insufferably smug having seen it in Hungary ad knew how to pronounce its name (Al Kath- oh-ee). John is in favour of calling the bat Alcathoe Bat rather than Alcathoe’s John kindly sent us a copy of his draft paper. We forwarded this to taxonomy keeny Derek Smith and we could almost hear him weeping as he e mailed us back post-haste “Hi Bob and Jude, many thanks for the paper. -

Index of Handbook of the Mammals of the World. Vol. 9. Bats
Index of Handbook of the Mammals of the World. Vol. 9. Bats A agnella, Kerivoula 901 Anchieta’s Bat 814 aquilus, Glischropus 763 Aba Leaf-nosed Bat 247 aladdin, Pipistrellus pipistrellus 771 Anchieta’s Broad-faced Fruit Bat 94 aquilus, Platyrrhinus 567 Aba Roundleaf Bat 247 alascensis, Myotis lucifugus 927 Anchieta’s Pipistrelle 814 Arabian Barbastelle 861 abae, Hipposideros 247 alaschanicus, Hypsugo 810 anchietae, Plerotes 94 Arabian Horseshoe Bat 296 abae, Rhinolophus fumigatus 290 Alashanian Pipistrelle 810 ancricola, Myotis 957 Arabian Mouse-tailed Bat 164, 170, 176 abbotti, Myotis hasseltii 970 alba, Ectophylla 466, 480, 569 Andaman Horseshoe Bat 314 Arabian Pipistrelle 810 abditum, Megaderma spasma 191 albatus, Myopterus daubentonii 663 Andaman Intermediate Horseshoe Arabian Trident Bat 229 Abo Bat 725, 832 Alberico’s Broad-nosed Bat 565 Bat 321 Arabian Trident Leaf-nosed Bat 229 Abo Butterfly Bat 725, 832 albericoi, Platyrrhinus 565 andamanensis, Rhinolophus 321 arabica, Asellia 229 abramus, Pipistrellus 777 albescens, Myotis 940 Andean Fruit Bat 547 arabicus, Hypsugo 810 abrasus, Cynomops 604, 640 albicollis, Megaerops 64 Andersen’s Bare-backed Fruit Bat 109 arabicus, Rousettus aegyptiacus 87 Abruzzi’s Wrinkle-lipped Bat 645 albipinnis, Taphozous longimanus 353 Andersen’s Flying Fox 158 arabium, Rhinopoma cystops 176 Abyssinian Horseshoe Bat 290 albiventer, Nyctimene 36, 118 Andersen’s Fruit-eating Bat 578 Arafura Large-footed Bat 969 Acerodon albiventris, Noctilio 405, 411 Andersen’s Leaf-nosed Bat 254 Arata Yellow-shouldered Bat 543 Sulawesi 134 albofuscus, Scotoecus 762 Andersen’s Little Fruit-eating Bat 578 Arata-Thomas Yellow-shouldered Talaud 134 alboguttata, Glauconycteris 833 Andersen’s Naked-backed Fruit Bat 109 Bat 543 Acerodon 134 albus, Diclidurus 339, 367 Andersen’s Roundleaf Bat 254 aratathomasi, Sturnira 543 Acerodon mackloti (see A. -

Chiropterology Division BC Arizona Trial Event 1 1. DESCRIPTION: Participants Will Be Assessed on Their Knowledge of Bats, With
Chiropterology Division BC Arizona Trial Event 1. DESCRIPTION: Participants will be assessed on their knowledge of bats, with an emphasis on North American Bats, South American Microbats, and African MegaBats. A TEAM OF UP TO: 2 APPROXIMATE TIME: 50 minutes 2. EVENT PARAMETERS: a. Each team may bring one 2” or smaller three-ring binder, as measured by the interior diameter of the rings, containing information in any form and from any source. Sheet protectors, lamination, tabs and labels are permitted in the binder. b. If the event features a rotation through a series of stations where the participants interact with samples, specimens or displays; no material may be removed from the binder throughout the event. c. In addition to the binder, each team may bring one unmodified and unannotated copy of either the National Bat List or an Official State Bat list which does not have to be secured in the binder. 3. THE COMPETITION: a. The competition may be run as timed stations and/or as timed slides/PowerPoint presentation. b. Specimens/Pictures will be lettered or numbered at each station. The event may include preserved specimens, skeletal material, and slides or pictures of specimens. c. Each team will be given an answer sheet on which they will record answers to each question. d. No more than 50% of the competition will require giving common or scientific names. e. Participants should be able to do a basic identification to the level indicated on the Official List. States may have a modified or regional list. See your state website. -

Threatened Jott
Journal ofThreatened JoTT TaxaBuilding evidence for conservation globally PLATINUM OPEN ACCESS 10.11609/jott.2020.12.3.15279-15406 www.threatenedtaxa.org 26 February 2020 (Online & Print) Vol. 12 | No. 3 | Pages: 15279–15406 ISSN 0974-7907 (Online) ISSN 0974-7893 (Print) ISSN 0974-7907 (Online); ISSN 0974-7893 (Print) Publisher Host Wildlife Information Liaison Development Society Zoo Outreach Organization www.wild.zooreach.org www.zooreach.org No. 12, Thiruvannamalai Nagar, Saravanampatti - Kalapatti Road, Saravanampatti, Coimbatore, Tamil Nadu 641035, India Ph: +91 9385339863 | www.threatenedtaxa.org Email: [email protected] EDITORS English Editors Mrs. Mira Bhojwani, Pune, India Founder & Chief Editor Dr. Fred Pluthero, Toronto, Canada Dr. Sanjay Molur Mr. P. Ilangovan, Chennai, India Wildlife Information Liaison Development (WILD) Society & Zoo Outreach Organization (ZOO), 12 Thiruvannamalai Nagar, Saravanampatti, Coimbatore, Tamil Nadu 641035, Web Design India Mrs. Latha G. Ravikumar, ZOO/WILD, Coimbatore, India Deputy Chief Editor Typesetting Dr. Neelesh Dahanukar Indian Institute of Science Education and Research (IISER), Pune, Maharashtra, India Mr. Arul Jagadish, ZOO, Coimbatore, India Mrs. Radhika, ZOO, Coimbatore, India Managing Editor Mrs. Geetha, ZOO, Coimbatore India Mr. B. Ravichandran, WILD/ZOO, Coimbatore, India Mr. Ravindran, ZOO, Coimbatore India Associate Editors Fundraising/Communications Dr. B.A. Daniel, ZOO/WILD, Coimbatore, Tamil Nadu 641035, India Mrs. Payal B. Molur, Coimbatore, India Dr. Mandar Paingankar, Department of Zoology, Government Science College Gadchiroli, Chamorshi Road, Gadchiroli, Maharashtra 442605, India Dr. Ulrike Streicher, Wildlife Veterinarian, Eugene, Oregon, USA Editors/Reviewers Ms. Priyanka Iyer, ZOO/WILD, Coimbatore, Tamil Nadu 641035, India Subject Editors 2016–2018 Fungi Editorial Board Ms. Sally Walker Dr. B. -
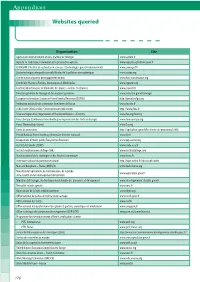
Appendices Websites Queried
Appendices Websites queried Organization Site Agence de l’environnement et de la maîtrise de l’énergie www.ademe.fr Agreste: la statistique, l’évaluation et la prospective agricole www.agreste.agriculture.gouv.fr CEMAGREF (l’Institut de recherche en sciences et technologies pour l’environnement) www.cemagref.fr Centre technique interprofessionnel d’études de la pollution atmosphérique www.citepa.org Comité national pour le développement du bois www.bois-construction.org Comité des Plantes à Parfum, Aromatiques et Médicinales www.cpparm.org Confédération française de l’industrie des papiers, cartons et celluloses www.copacel.fr Direction générale de l’énergie et des matières premières www.industrie.gouv.fr/energie European Information System on Forest Genetic Resources (EUFGIS) http://portal.eufgis.org Fédération nationale des communes forestières de France www.fncofor.fr FCBA (Forêt Cellulose Bois-Construction Ameublement) http://www.fcba.fr Food and Agriculture Organization of the United Nations – Forestry www.fao.org/forestry Forest Europe (Conférence ministérielle pour la protection des forêts en Europe) www.foresteurope.org Forest Stewardship Council www.fsc.org Forêts de protection http://agriculture.gouv.fr/les-forets-de-protection,10806 French National Forest Inventory (Inventaire forestier national) www.ifn.fr Groupement d’intérêt public Écosystèmes Forestiers www.gip-ecofor.org Institut de l’abeille (ITSAP) www.cnda.asso.fr Institut méditerranéen du liège (IML) www.institutduliege.com Institut national de la statistique et des -

Hungary and Slovakia, 2017
HUNGARY and SLOVAKIA SMALL MAMMAL TOUR - The Bats and Rodents of Central Europe Hangarian hay meadow in warm August sunshine. Steve Morgan ([email protected]), John Smart 25/8/17 HUNGARY and SLOVAKIA SMALL MAMMAL TOUR 1 Introduction I had long intended to visit Hungary for bats and small mammals but had never quite got round to it. Now, however, a chance presented itself to join a tour with both Hungary and Slovakia on the itinerary and a long list of prospective mammalian targets on offer, including Forest Dormouse, European Hamster, Lesser Mole Rat, Common Souslik and a number of highly desirable bats such as Grey Long-eared, Northern and Parti-coloured. The tour was organised by Ecotours of Hungary and led by Istvan Bartol. It ran from 9/8/17 to 17/8/17, the two particpants being John Smart and me, both of us from the UK. 2 Logistics I flew from Luton to Budapest on Wizzair. Frankly, I’d never heard of Wizzair before and, given their two hour delay on the outward leg (resulting in an extremely late check in to my hotel in Budapest), I’m not sure I want to hear about them again! The hotels selected by Ecotours were all very good. In Mezokovesd we stayed at the Hajnal Hotel which was clean and comfortable and offered a good (cooked) buffet breakfast. In Slovakia we stayed at the equally good Penzion Reva which was set in very nice countryside overlooking a picturesque lake. Istvan Bartol led the tour and did all the driving. -

The Evolution of Echolocation in Bats: a Comparative Approach
The evolution of echolocation in bats: a comparative approach Alanna Collen A thesis submitted for the degree of Doctor of Philosophy from the Department of Genetics, Evolution and Environment, University College London. November 2012 Declaration Declaration I, Alanna Collen (née Maltby), confirm that the work presented in this thesis is my own. Where information has been derived from other sources, this is indicated in the thesis, and below: Chapter 1 This chapter is published in the Handbook of Mammalian Vocalisations (Maltby, Jones, & Jones) as a first authored book chapter with Gareth Jones and Kate Jones. Gareth Jones provided the research for the genetics section, and both Kate Jones and Gareth Jones providing comments and edits. Chapter 2 The raw echolocation call recordings in EchoBank were largely made and contributed by members of the ‘Echolocation Call Consortium’ (see full list in Chapter 2). The R code for the diversity maps was provided by Kamran Safi. Custom adjustments were made to the computer program SonoBat by developer Joe Szewczak, Humboldt State University, in order to select echolocation calls for measurement. Chapter 3 The supertree construction process was carried out using Perl scripts developed and provided by Olaf Bininda-Emonds, University of Oldenburg, and the supertree was run and dated by Olaf Bininda-Emonds. The source trees for the Pteropodidae were collected by Imperial College London MSc student Christina Ravinet. Chapter 4 Rob Freckleton, University of Sheffield, and Luke Harmon, University of Idaho, helped with R code implementation. 2 Declaration Chapter 5 Luke Harmon, University of Idaho, helped with R code implementation. Chapter 6 Joseph W. -

Introduction to the Irish Vespertilionid Bats
A conservation plan for Irish vesper bats Irish Wildlife Manuals No. 20 A conservation plan for Irish vesper bats Kate McAney The Vincent Wildlife Trust Donaghpatrick Headford Co. Galway Citation: McAney, K. (2006) A conservation plan for Irish vesper bats. Irish Wildlife Manuals, No. 20. National Parks and Wildlife Service, Department of Environment, Heritage and Local Government, Dublin, Ireland. Cover photo: Leisler’s bat Nyctalus leisleri by Eddie Dunne (© NPWS) Irish Wildlife Manuals Series Editor: F. Marnell © National Parks and Wildlife Service 2006 ISSN 1393 – 6670 2 Summary ..................................................................................................................................... 4 1. Introduction ......................................................................................................................... 5 2. Species Descriptions............................................................................................................ 6 2.1 Introduction ................................................................................................................. 6 2.2 Common pipistrelle Pipistrellus pipistrellus (Schreber, 1774) &............................... 7 2.3 Nathusius’ pipistrelle Pipistrellus nathusii (Keyserling & Blasius, 1839) ............... 11 2.4 Whiskered bat Myotis mystacinus (Kuhl, 1817) & ................................................... 12 2.5 Brown long-eared bat Plecotus auritus (Linnaeus, 1758)......................................... 15 2.6 Natterer’s bat Myotis