Identification of the Fungal Catabolic D-Galacturonate Pathway
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Sulfite Dehydrogenases in Organotrophic Bacteria : Enzymes
Sulfite dehydrogenases in organotrophic bacteria: enzymes, genes and regulation. Dissertation zur Erlangung des akademischen Grades des Doktors der Naturwissenschaften (Dr. rer. nat.) an der Universität Konstanz Fachbereich Biologie vorgelegt von Sabine Lehmann Tag der mündlichen Prüfung: 10. April 2013 1. Referent: Prof. Dr. Bernhard Schink 2. Referent: Prof. Dr. Andrew W. B. Johnston So eine Arbeit wird eigentlich nie fertig, man muss sie für fertig erklären, wenn man nach Zeit und Umständen das möglichste getan hat. (Johann Wolfgang von Goethe, Italienische Reise, 1787) DANKSAGUNG An dieser Stelle möchte ich mich herzlich bei folgenden Personen bedanken: . Prof. Dr. Alasdair M. Cook (Universität Konstanz, Deutschland), der mir dieses Thema und seine Laboratorien zur Verfügung stellte, . Prof. Dr. Bernhard Schink (Universität Konstanz, Deutschland), für seine spontane und engagierte Übernahme der Betreuung, . Prof. Dr. Andrew W. B. Johnston (University of East Anglia, UK), für seine herzliche und bereitwillige Aufnahme in seiner Arbeitsgruppe, seiner engagierten Unter- stützung, sowie für die Übernahme des Koreferates, . Prof. Dr. Frithjof C. Küpper (University of Aberdeen, UK), für seine große Hilfsbereitschaft bei der vorliegenden Arbeit und geplanter Manuskripte, als auch für die mentale Unterstützung während der letzten Jahre! Desweiteren möchte ich herzlichst Dr. David Schleheck für die Übernahme des Koreferates der mündlichen Prüfung sowie Prof. Dr. Alexander Bürkle, für die Übernahme des Prüfungsvorsitzes sowie für seine vielen hilfreichen Ratschläge danken! Ein herzliches Dankeschön geht an alle beteiligten Arbeitsgruppen der Universität Konstanz, der UEA und des SAMS, ganz besonders möchte ich dabei folgenden Personen danken: . Dr. David Schleheck und Karin Denger, für die kritische Durchsicht dieser Arbeit, der durch und durch sehr engagierten Hilfsbereitschaft bei Problemen, den zahlreichen wissenschaftlichen Diskussionen und für die aufbauenden Worte, . -

Assembly of an Active Enzyme by the Linkage of Two Protein Modules
Proc. Natl. Acad. Sci. USA Vol. 94, pp. 1069–1073, February 1997 Biochemistry Assembly of an active enzyme by the linkage of two protein modules A. E. NIXON,M.S.WARREN, AND S. J. BENKOVIC* 152 Davey Laboratory, Department of Chemistry, Pennsylvania State University, University Park, PA 16802-6300 Contributed by S. J. Benkovic, December 9, 1996 ABSTRACT The feasibility of creating new enzyme activ- design enzymes with novel properties. Previous approaches to ities from enzymes of known function has precedence in view the design of proteins with novel activities have included of protein evolution based on the concepts of molecular catalytic antibodies (8, 9); introduction of metal ion binding recruitment and exon shuffling. The enzymes encoded by the sites, such as the one engineered into trypsin to allow either Escherichia coli genes purU and purN, N10-formyltetrahydro- control of the proteolytic activity (10) or to regulate specificity folate hydrolase and glycinamide ribonucleotide (GAR) trans- (11); creation of hybrid enzymes through exchange of subunits formylase, respectively, catalyze similiar yet distinct reactions. to create hybrid oligomers (12); replacement of structural N10-formyltetrahydrofolate hydrolase uses water to cleave elements such as the DNA binding domain of GCN4 with that N10-formyltetrahydrofolate into tetrahydrofolate and for- of CyEBP (13); mutation of multiple individual residues to mate, whereas GAR transformylase catalyses the transfer of change the cofactor specificity of glutathione reductase from formyl from N10-formyltetrahydrofolate to GAR to yield NADPH to NADH (14), modulation of the substrate speci- formyl-GAR and tetrahydrofolate. The two enzymes show ficity of aspartate aminotransferase (15); and changing the significant homology ('60%) in the carboxyl-terminal region specificity of subtilisin (16) and a-lytic protease (17) through which, from the GAR transformylase crystal structure and mutation of single functional groups. -

Resolution of Carbon Metabolism and Sulfur-Oxidation Pathways of Metallosphaera Cuprina Ar-4 Via Comparative Proteomics
JOURNAL OF PROTEOMICS 109 (2014) 276– 289 Available online at www.sciencedirect.com ScienceDirect www.elsevier.com/locate/jprot Resolution of carbon metabolism and sulfur-oxidation pathways of Metallosphaera cuprina Ar-4 via comparative proteomics Cheng-Ying Jianga, Li-Jun Liua, Xu Guoa, Xiao-Yan Youa, Shuang-Jiang Liua,c,⁎, Ansgar Poetschb,⁎⁎ aState Key Laboratory of Microbial Resources, Institute of Microbiology, Chinese Academy of Sciences, Beijing, PR China bPlant Biochemistry, Ruhr University Bochum, Bochum, Germany cEnvrionmental Microbiology and Biotechnology Research Center, Institute of Microbiology, Chinese Academy of Sciences, Beijing, PR China ARTICLE INFO ABSTRACT Article history: Metallosphaera cuprina is able to grow either heterotrophically on organics or autotrophically Received 16 March 2014 on CO2 with reduced sulfur compounds as electron donor. These traits endowed the species Accepted 6 July 2014 desirable for application in biomining. In order to obtain a global overview of physiological Available online 14 July 2014 adaptations on the proteome level, proteomes of cytoplasmic and membrane fractions from cells grown autotrophically on CO2 plus sulfur or heterotrophically on yeast extract Keywords: were compared. 169 proteins were found to change their abundance depending on growth Quantitative proteomics condition. The proteins with increased abundance under autotrophic growth displayed Bioleaching candidate enzymes/proteins of M. cuprina for fixing CO2 through the previously identified Autotrophy 3-hydroxypropionate/4-hydroxybutyrate cycle and for oxidizing elemental sulfur as energy Heterotrophy source. The main enzymes/proteins involved in semi- and non-phosphorylating Entner– Industrial microbiology Doudoroff (ED) pathway and TCA cycle were less abundant under autotrophic growth. Also Extremophile some transporter proteins and proteins of amino acid metabolism changed their abundances, suggesting pivotal roles for growth under the respective conditions. -

Dissimilation of Cysteate Via 3-Sulfolactate Sulfo-Lyase and a Sulfate Exporter in Paracoccus Pantotrophus NKNCYSA
Microbiology (2005), 151, 737–747 DOI 10.1099/mic.0.27548-0 Dissimilation of cysteate via 3-sulfolactate sulfo-lyase and a sulfate exporter in Paracoccus pantotrophus NKNCYSA Ulrike Rein,1 Ronnie Gueta,1 Karin Denger,1 Ju¨rgen Ruff,1 Klaus Hollemeyer2 and Alasdair M. Cook1 Correspondence 1Department of Biology, The University, D-78457 Konstanz, Germany Alasdair Cook 2Institute of Biochemical Engineering, Saarland University, Box 50 11 50, D-66041 [email protected] Saarbru¨cken, Germany Paracoccus pantotrophus NKNCYSA utilizes (R)-cysteate (2-amino-3-sulfopropionate) as a sole source of carbon and energy for growth, with either nitrate or molecular oxygen as terminal electron acceptor, and the specific utilization rate of cysteate is about 2 mkat (kg protein)”1. The initial degradative reaction is catalysed by an (R)-cysteate : 2-oxoglutarate aminotransferase, which yields 3-sulfopyruvate. The latter was reduced to 3-sulfolactate by an NAD-linked sulfolactate dehydrogenase [3?3 mkat (kg protein)”1]. The inducible desulfonation reaction was not detected initially in cell extracts. However, a strongly induced protein with subunits of 8 kDa (a) and 42 kDa (b) was found and purified. The corresponding genes had similarities to those encoding altronate dehydratases, which often require iron for activity. The purified enzyme could then be shown to convert 3-sulfolactate to sulfite and pyruvate and it was termed sulfolactate sulfo-lyase (Suy). A high level of sulfite dehydrogenase was also induced during growth with cysteate, and the organism excreted sulfate. A putative regulator, OrfR, was encoded upstream of suyAB on the reverse strand. Downstream of suyAB was suyZ, which was cotranscribed with suyB. -
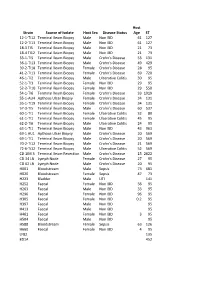
Strain Source of Isolate Host Sex Disease Status Host Age ST
Host Strain Source of Isolate Host Sex Disease Status Age ST 12-1-TI12 Terminal Ileum Biopsy Male Non IBD 61 127 12-2-TI13 Terminal Ileum Biopsy Male Non IBD 61 127 18-3 TI5 Terminal Ileum Biopsy Male Non IBD 21 73 18-4 TI12 Terminal Ileum Biopsy Male Non IBD 21 73 33-1-TI5 Terminal Ileum Biopsy Male Crohn's Disease 53 131 36-1-TI13 Terminal Ileum Biopsy Male Crohn's Disease 49 429 39-2-TI18 Terminal Ileum Biopsy Female Crohn's Disease 28 95 41-2-TI13 Terminal Ileum Biopsy Female Crohn's Disease 69 720 46-1-TI2 Terminal Ileum Biopsy Male Ulcerative Colitis 30 95 52-1-TI3 Terminal Ileum Biopsy Female Non IBD 29 95 52-2-TI10 Terminal Ileum Biopsy Female Non IBD 29 550 54-1-TI6 Terminal Ileum Biopsy Female Crohn's Disease 33 1919 55-1-AU4 Apthous Ulcer Biopsy Female Crohn's Disease 34 131 55-1-TI19 Terminal Ileum Biopsy Female Crohn's Disease 34 131 57-3-TI5 Terminal Ileum Biopsy Male Crohn's Disease 60 537 60-1-TI1 Terminal Ileum Biopsy Female Ulcerative Colitis 32 80 61-1-TI1 Terminal Ileum Biopsy Female Ulcerative Colitis 45 95 62-2-TI6 Terminal Ileum Biopsy Male Ulcerative Colitis 24 95 63-1-TI1 Terminal Ileum Biopsy Male Non IBD 43 963 69-1 AU1 Apthous Ulcer Biopsy Male Crohn's Disease 20 569 69-1-TI1 Terminal Ileum Biopsy Male Crohn's Disease 20 569 70-2-TI12 Terminal Ileum Biopsy Male Crohn's Disease 21 569 72-6-Ti12 Terminal Ileum Biopsy Male Ulcerative Colitis 52 569 CD 1IM 3 Terminal Ileum Resection Male Crohn's Disease 15 2622 CD 34 LN Lymph Node Female Crohn's Disease 27 95 CD 62 LN Lymph Node Male Crohn's Disease 20 95 H001 -

Supplementary Information
Supplementary information (a) (b) Figure S1. Resistant (a) and sensitive (b) gene scores plotted against subsystems involved in cell regulation. The small circles represent the individual hits and the large circles represent the mean of each subsystem. Each individual score signifies the mean of 12 trials – three biological and four technical. The p-value was calculated as a two-tailed t-test and significance was determined using the Benjamini-Hochberg procedure; false discovery rate was selected to be 0.1. Plots constructed using Pathway Tools, Omics Dashboard. Figure S2. Connectivity map displaying the predicted functional associations between the silver-resistant gene hits; disconnected gene hits not shown. The thicknesses of the lines indicate the degree of confidence prediction for the given interaction, based on fusion, co-occurrence, experimental and co-expression data. Figure produced using STRING (version 10.5) and a medium confidence score (approximate probability) of 0.4. Figure S3. Connectivity map displaying the predicted functional associations between the silver-sensitive gene hits; disconnected gene hits not shown. The thicknesses of the lines indicate the degree of confidence prediction for the given interaction, based on fusion, co-occurrence, experimental and co-expression data. Figure produced using STRING (version 10.5) and a medium confidence score (approximate probability) of 0.4. Figure S4. Metabolic overview of the pathways in Escherichia coli. The pathways involved in silver-resistance are coloured according to respective normalized score. Each individual score represents the mean of 12 trials – three biological and four technical. Amino acid – upward pointing triangle, carbohydrate – square, proteins – diamond, purines – vertical ellipse, cofactor – downward pointing triangle, tRNA – tee, and other – circle. -

Supplementary Informations SI2. Supplementary Table 1
Supplementary Informations SI2. Supplementary Table 1. M9, soil, and rhizosphere media composition. LB in Compound Name Exchange Reaction LB in soil LBin M9 rhizosphere H2O EX_cpd00001_e0 -15 -15 -10 O2 EX_cpd00007_e0 -15 -15 -10 Phosphate EX_cpd00009_e0 -15 -15 -10 CO2 EX_cpd00011_e0 -15 -15 0 Ammonia EX_cpd00013_e0 -7.5 -7.5 -10 L-glutamate EX_cpd00023_e0 0 -0.0283302 0 D-glucose EX_cpd00027_e0 -0.61972444 -0.04098397 0 Mn2 EX_cpd00030_e0 -15 -15 -10 Glycine EX_cpd00033_e0 -0.0068175 -0.00693094 0 Zn2 EX_cpd00034_e0 -15 -15 -10 L-alanine EX_cpd00035_e0 -0.02780553 -0.00823049 0 Succinate EX_cpd00036_e0 -0.0056245 -0.12240603 0 L-lysine EX_cpd00039_e0 0 -10 0 L-aspartate EX_cpd00041_e0 0 -0.03205557 0 Sulfate EX_cpd00048_e0 -15 -15 -10 L-arginine EX_cpd00051_e0 -0.0068175 -0.00948672 0 L-serine EX_cpd00054_e0 0 -0.01004986 0 Cu2+ EX_cpd00058_e0 -15 -15 -10 Ca2+ EX_cpd00063_e0 -15 -100 -10 L-ornithine EX_cpd00064_e0 -0.0068175 -0.00831712 0 H+ EX_cpd00067_e0 -15 -15 -10 L-tyrosine EX_cpd00069_e0 -0.0068175 -0.00233919 0 Sucrose EX_cpd00076_e0 0 -0.02049199 0 L-cysteine EX_cpd00084_e0 -0.0068175 0 0 Cl- EX_cpd00099_e0 -15 -15 -10 Glycerol EX_cpd00100_e0 0 0 -10 Biotin EX_cpd00104_e0 -15 -15 0 D-ribose EX_cpd00105_e0 -0.01862144 0 0 L-leucine EX_cpd00107_e0 -0.03596182 -0.00303228 0 D-galactose EX_cpd00108_e0 -0.25290619 -0.18317325 0 L-histidine EX_cpd00119_e0 -0.0068175 -0.00506825 0 L-proline EX_cpd00129_e0 -0.01102953 0 0 L-malate EX_cpd00130_e0 -0.03649016 -0.79413596 0 D-mannose EX_cpd00138_e0 -0.2540567 -0.05436649 0 Co2 EX_cpd00149_e0 -

Supplementary Information
Supplementary Information Laboratory cultivation of acidophilic nanoorganisms. Physiological and bioinformatic dissection of a stable laboratory co-culture. Susanne Krause [1], Andreas Bremges [3,4], Philipp C. Münch [3,5], Alice C. McHardy [3] and Johannes Gescher* [1,2] [1] Department of Applied Biology, Karlsruhe Institute of Technology (KIT), Karlsruhe, Germany [2] Institute for Biological Interfaces, Karlsruhe Institute of Technology (KIT), Eggenstein- Leopoldshafen, Germany [3] Computational Biology of Infection Research, Helmholtz Centre for Infection Research, Braunschweig, Germany [4] German Center for Infection Research (DZIF), partner site Hannover-Braunschweig, Braunschweig, Germany [5] Max von Pettenkofer-Institute of Hygiene and Medical Microbiology, Ludwig-Maximilians- University of Munich, Munich, Germany SI Materials and Methods Quantification of cells using quantitative PCR The target sequences were amplified by PCR from genomic DNA of the enrichment cultures using primers see table S8. The amplified fragments contained overlapping regions as well as a BamHI site at the 5’ end and a SacI site at the 3’ end. Using the overlaps, the fragments were combined and cloned via the added restrictions sites into plasmid pAH95 1. Integration in the genome of E. coli DH5alphaZ1 was conducted as described before 1. For standard curve design, serial dilutions of E. coli DH5alphaZ1 cells containing the merged 23S target sequences of the ARMAN und Thermoplasmatales enrichments were prepared and cells were counted in a Neubauer counting chamber (Marienfeld, Lauda-Königshofen, Germany). DNA of the dilution series as well as of enrichment-cultures was extracted using the innuSPEED soil DNA kit (Analytic Jena, Jena, Germany) with minor modifications to the manufacturer´s instructions. -

Novel Non-Phosphorylative Pathway of Pentose Metabolism from Bacteria
www.nature.com/scientificreports OPEN Novel non-phosphorylative pathway of pentose metabolism from bacteria Received: 22 March 2018 Seiya Watanabe1,2,3, Fumiyasu Fukumori4, Hisashi Nishiwaki1,2, Yasuhiro Sakurai5, Accepted: 30 September 2018 Kunihiko Tajima5 & Yasuo Watanabe1,2 Published: xx xx xxxx Pentoses, including D-xylose, L-arabinose, and D-arabinose, are generally phosphorylated to D-xylulose 5-phosphate in bacteria and fungi. However, in non-phosphorylative pathways analogous to the Entner-Dodorof pathway in bacteria and archaea, such pentoses can be converted to pyruvate and glycolaldehyde (Route I) or α-ketoglutarate (Route II) via a 2-keto-3-deoxypentonate (KDP) intermediate. Putative gene clusters related to these metabolic pathways were identifed on the genome of Herbaspirillum huttiense IAM 15032 using a bioinformatic analysis. The biochemical characterization of C785_RS13685, one of the components encoded to D-arabinonate dehydratase, difered from the known acid-sugar dehydratases. The biochemical characterization of the remaining components and a genetic expression analysis revealed that D- and L-KDP were converted not only to α-ketoglutarate, but also pyruvate and glycolate through the participation of dehydrogenase and hydrolase (Route III). Further analyses revealed that the Route II pathway of D-arabinose metabolism was not evolutionally related to the analogous pathway from archaea. Te breakdown of D-glucose is central for energy and biosynthetic metabolism throughout all domains of life. Te most common glycolytic routes in bacteria are the Embden-Meyerhof-Parnas, the Entner-Doudorof (ED), and the oxidative pentose phosphate pathways. Te distinguishing diference between the two former glyco- lytic pathways lies in the nature of the 6-carbon metabolic intermediates that serve as substrates for aldol cleav- age. -

Within-Host Diversity of Multidrug-Resistant Escherichia Coli in the Gut and Bladder Sofiya G. Shevchenko a Dissertation Submitt
Within-host Diversity of Multidrug-Resistant Escherichia coli in the Gut and Bladder Sofiya G. Shevchenko A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy University of Washington 2019 Reading committee: Evgeni Sokurenko, Chair John Mittler Kevin Hybiske Program Authorized to Offer Degree: Microbiology ©Copyright 2019 Sofiya G. Shevchenko University of Washington Abstract Within-host Diversity of Escherichia coli in the Gut and Bladder Sofiya G. Shevchenko Chair of the Supervisory Committee: Evgeni V. Sokurenko Department of Microbiology Uropathogenic E. coli are paradoxically able to both cause disease in the urinary tract, and reside there asymptomatically. The pandemic, multi-drug resistant E. coli subclone ST131-H30 (H30) is of special interest, as it has been found to persist in the gut and bladder of healthy people. In order to understand this persistence, we investigated whether H30 is competitive in these niches and thus able to persist by excluding other E. coli, as well as whether H30 may persist via within-host adaptation. In order to assess the E. coli clonal landscape, we developed a novel method based on deep sequencing of two loci, along with an algorithm for analysis of resulting data. Using this method, we assessed fecal and urinary samples from healthy women carrying H30, and found that even in the absence of antibiotic use, H30 could completely dominate the gut and, especially, urine of healthy carriers. In order to ascertain whether H30 adapts within- host, we employed population-level whole genome sequencing, and determined that H30 undergoes changes in genes affecting respiration in the gut, similar to commensal gut E. -

8-Barrel Enzymes John a Gerlt� and Frank M Raushely
252 Evolution of function in (b/a)8-barrel enzymes John A Gerltà and Frank M Raushely The (b/a)8-barrel is the most common fold in structurally closed, parallel b-sheet structure of the (b/a)8-barrel is characterized enzymes. Whether the functionally diverse formed from eight parallel (b/a)-units linked by hydrogen enzymes that share this fold are the products of either divergent bonds that form a cylindrical core. Despite its eightfold or convergent evolution (or both) is an unresolved question that pseudosymmetry, the packing within the interior of the will probably be answered as the sequence databases continue barrel is better described as four (b/a)2-subdomains in to expand. Recent work has examined natural, designed, and which distinct hydrophobic cores are located between the directed evolution of function in several superfamilies of (b/a)8- b-sheets and flanking a-helices [2]. barrel containing enzymes. The active sites are located at the C-terminal ends of the Addresses b-strands. So placed, the functional groups surround the ÃDepartments of Biochemistry and Chemistry, University of Illinois at active site and are structurally independent: the ‘old’ and Urbana-Champaign, 600 South Mathews Avenue, Urbana, ‘new’ enzymes retain functional groups at the ends of Illinois 61801, USA e-mail: [email protected] some b-strands, but others are altered to allow the ‘new’ yDepartment of Chemistry, Texas A&M University, PO Box 30012, activity [3]. With this blueprint, the (b/a)8-barrel is opti- College Station, Texas 77842-3012, USA mized for evolution of new functions. -

Supplementary Material
Supplementary material Figure S1. Cluster analysis of the proteome profile based on qualitative data in low and high sugar conditions. Figure S2. Expression pattern of proteins under high and low sugar cultivation of Granulicella sp. WH15 a) All proteins identified in at least two out of three replicates (excluding on/off proteins). b) Only proteins with significant change t-test p=0.01. 2fold change is indicated by a red line. Figure S3. TigrFam roles of the differentially expressed proteins, excluding proteins with unknown function. Figure S4. General overview of up (red) and downregulated (blue) metabolic pathways based on KEGG analysis of proteome. Table S1. growth of strain Granulicella sp. WH15 in culture media supplemented with different carbon sources. Carbon Source Growth Pectin - Glycogen - Glucosamine - Cellulose - D-glucose + D-galactose + D-mannose + D-xylose + L-arabinose + L-rhamnose + D-galacturonic acid - Cellobiose + D-lactose + Sucrose + +=positive growth; -=No growth. Table S2. Total number of transcripts reads per sample in low and high sugar conditions. Sample ID Total Number of Reads Low sugar (1) 15,731,147 Low sugar (2) 12,624,878 Low sugar (3) 11,080,985 High sugar (1) 11,138,128 High sugar (2) 9,322,795 High sugar (3) 10,071,593 Table S3. Differentially up and down regulated transcripts in high sugar treatment. ORF Annotation Log2FC GWH15_14040 hypothetical protein 3.71 GWH15_06005 hypothetical protein 3.12 GWH15_00285 tRNA-Asn(gtt) 2.74 GWH15_06010 hypothetical protein 2.70 GWH15_14055 hypothetical protein 2.66