Development of Rapid and Inexpensive Derivatisation Methods for Methylphosphonic Acid
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Reaction Kinetics of the Alcoholysis of Substituted Benzoyl Chlorides
Proceedings of the Iowa Academy of Science Volume 61 Annual Issue Article 26 1954 Reaction Kinetics of the Alcoholysis of Substituted Benzoyl Chlorides B. R. Bluestein Coe College Albert Hybl Coe College Yoshimi Al Nishioka Coe College Let us know how access to this document benefits ouy Copyright ©1954 Iowa Academy of Science, Inc. Follow this and additional works at: https://scholarworks.uni.edu/pias Recommended Citation Bluestein, B. R.; Hybl, Albert; and Nishioka, Yoshimi Al (1954) "Reaction Kinetics of the Alcoholysis of Substituted Benzoyl Chlorides," Proceedings of the Iowa Academy of Science, 61(1), 225-232. Available at: https://scholarworks.uni.edu/pias/vol61/iss1/26 This Research is brought to you for free and open access by the Iowa Academy of Science at UNI ScholarWorks. It has been accepted for inclusion in Proceedings of the Iowa Academy of Science by an authorized editor of UNI ScholarWorks. For more information, please contact [email protected]. Bluestein et al.: Reaction Kinetics of the Alcoholysis of Substituted Benzoyl Chlor Reaction Kinetics of the Alcoholysis of Substituted Benzoyl Chlorides By B. R. BLUESTEIN, ALBERT HYBL* AND YosHIMI AL NISHIOKA INTRODUCTION The reaction kinetics of the alcoholysis of substituted benzoyl chlorides was studied. The mechanism of the alcoholysis reaction, which is most generally accepted ( 1), shows that the overall re action should be second-order and that the reaction should be first-order with respect to the acid chloride and first-order with respect to the alcohol. This rate study was carried out using a large excess of alcohol as the solvent, thus obtaining pseudo-first order rate constants, first-order with respect to the acid chloride only. -

Solvent-Free and Safe Process for the Quantitative Production of Phosgene from Triphosgene by Deactivated Imino-Based Catalysts
Organic Process Research & Development 2010, 14, 1501–1505 Solvent-Free and Safe Process for the Quantitative Production of Phosgene from Triphosgene by Deactivated Imino-Based Catalysts Heiner Eckert* and Johann Auerweck Department of Chemistry, Technische UniVersitaet Muenchen, Lichtenbergstr. 4, Garching 85747, Germany Abstract: Scheme 1. Decomposition of triphosgene (1a) into carbon tetrachloride, carbon dioxide, and 1 equiv of phosgene (3) Phosgene is quantitatively formed from solid triphosgene in a solvent-free and safe process without any reaction heat, catalyzed by planar N-heterocycles with deactivated imino functions. The rate of phosgene generation is adjustable to the rate of phosgene consumption in the subsequent phosgenation reaction by thermal control, catalyst concentration, and in some cases, specific proper- ties of selected metal phthalocyanines. A thermal runaway reaction of this process is impossible. phosgene in most reactions, yet several phosgenation reactions are advantageously carried out with phosgene, that is, when excessive triphosgene is difficult to remove during the reaction Introduction workup because of its high boiling point of over 200 °C. Its Phosgene (3) is a highly useful and versatile chemical in excess can be destroyed by hydrolysis, when phosgenation performing syntheses.1a Although consisting of only four atoms, products are not sensitive to moisture as are carbonates, four important transformations can be carried out with it in carbamates, ureas, diarylketones, alkylhalides, cyanides, and organic -

Toxicological Profile for Glyphosate Were
A f Toxicological Profile for Glyphosate August 2020 GLYPHOSATE II DISCLAIMER Use of trade names is for identification only and does not imply endorsement by the Agency for Toxic Substances and Disease Registry, the Public Health Service, or the U.S. Department of Health and Human Services. GLYPHOSATE III FOREWORD This toxicological profile is prepared in accordance with guidelines developed by the Agency for Toxic Substances and Disease Registry (ATSDR) and the Environmental Protection Agency (EPA). The original guidelines were published in the Federal Register on April 17, 1987. Each profile will be revised and republished as necessary. The ATSDR toxicological profile succinctly characterizes the toxicologic and adverse health effects information for these toxic substances described therein. Each peer-reviewed profile identifies and reviews the key literature that describes a substance's toxicologic properties. Other pertinent literature is also presented, but is described in less detail than the key studies. The profile is not intended to be an exhaustive document; however, more comprehensive sources of specialty information are referenced. The focus of the profiles is on health and toxicologic information; therefore, each toxicological profile begins with a relevance to public health discussion which would allow a public health professional to make a real-time determination of whether the presence of a particular substance in the environment poses a potential threat to human health. The adequacy of information to determine a substance's -

Efficient Esterification of Oxidized L-Glutathione and Other Small Peptides
Molecules 2015, 20, 10487-10495; doi:10.3390/molecules200610487 OPEN ACCESS molecules ISSN 1420-3049 www.mdpi.com/journal/molecules Communication Efficient Esterification of Oxidized L-Glutathione and Other Small Peptides Emily R. Vogel, William Jackson and Douglas S. Masterson * Department of Chemistry and Biochemistry, the University of Southern Mississippi, 118 College Drive #5043, Hattiesburg, MS 39406, USA; E-Mails: [email protected] (E.R.V.); [email protected] (W.J.) * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel.: +1-601-266-4714. Academic Editor: Derek J. McPhee Received: 17 May 2015 / Accepted: 4 June 2015 / Published: 8 June 2015 Abstract: Oxidized L-glutathione was esterified to the tetra methyl ester using thionyl chloride in methanol solvent. Other alcohols were tested and the reaction progress was monitored via ESI-MS. This procedure proved to be compatible with other small peptides not containing serine and cysteine residues. In contrast to previously reported methods this procedure provided convenient access to esterified peptides requiring no purification, extended reaction times, or complicated reaction setups. Keywords: esterification; peptide esterification; amino acids; carboxylic acids; esters 1. Introduction The design and synthesis of novel glutathione analogues are studied extensively for their pharmacological properties in the treatment of a wide range of diseases [1–3]. Reduced glutathione (GSH), -L-glutamyl-L-cysteinyl-glycine, is prone to reactivity at the sulfhydryl, terminal amino group, and both carbonyls. Thus, the synthesis of GSH analogues are a synthetic challenge to chemists, and the clever manipulation of protecting groups are needed to prevent unwanted reactions. -

Chemical Name Federal P Code CAS Registry Number Acutely
Acutely / Extremely Hazardous Waste List Federal P CAS Registry Acutely / Extremely Chemical Name Code Number Hazardous 4,7-Methano-1H-indene, 1,4,5,6,7,8,8-heptachloro-3a,4,7,7a-tetrahydro- P059 76-44-8 Acutely Hazardous 6,9-Methano-2,4,3-benzodioxathiepin, 6,7,8,9,10,10- hexachloro-1,5,5a,6,9,9a-hexahydro-, 3-oxide P050 115-29-7 Acutely Hazardous Methanimidamide, N,N-dimethyl-N'-[2-methyl-4-[[(methylamino)carbonyl]oxy]phenyl]- P197 17702-57-7 Acutely Hazardous 1-(o-Chlorophenyl)thiourea P026 5344-82-1 Acutely Hazardous 1-(o-Chlorophenyl)thiourea 5344-82-1 Extremely Hazardous 1,1,1-Trichloro-2, -bis(p-methoxyphenyl)ethane Extremely Hazardous 1,1a,2,2,3,3a,4,5,5,5a,5b,6-Dodecachlorooctahydro-1,3,4-metheno-1H-cyclobuta (cd) pentalene, Dechlorane Extremely Hazardous 1,1a,3,3a,4,5,5,5a,5b,6-Decachloro--octahydro-1,2,4-metheno-2H-cyclobuta (cd) pentalen-2- one, chlorecone Extremely Hazardous 1,1-Dimethylhydrazine 57-14-7 Extremely Hazardous 1,2,3,4,10,10-Hexachloro-6,7-epoxy-1,4,4,4a,5,6,7,8,8a-octahydro-1,4-endo-endo-5,8- dimethanonaph-thalene Extremely Hazardous 1,2,3-Propanetriol, trinitrate P081 55-63-0 Acutely Hazardous 1,2,3-Propanetriol, trinitrate 55-63-0 Extremely Hazardous 1,2,4,5,6,7,8,8-Octachloro-4,7-methano-3a,4,7,7a-tetra- hydro- indane Extremely Hazardous 1,2-Benzenediol, 4-[1-hydroxy-2-(methylamino)ethyl]- 51-43-4 Extremely Hazardous 1,2-Benzenediol, 4-[1-hydroxy-2-(methylamino)ethyl]-, P042 51-43-4 Acutely Hazardous 1,2-Dibromo-3-chloropropane 96-12-8 Extremely Hazardous 1,2-Propylenimine P067 75-55-8 Acutely Hazardous 1,2-Propylenimine 75-55-8 Extremely Hazardous 1,3,4,5,6,7,8,8-Octachloro-1,3,3a,4,7,7a-hexahydro-4,7-methanoisobenzofuran Extremely Hazardous 1,3-Dithiolane-2-carboxaldehyde, 2,4-dimethyl-, O- [(methylamino)-carbonyl]oxime 26419-73-8 Extremely Hazardous 1,3-Dithiolane-2-carboxaldehyde, 2,4-dimethyl-, O- [(methylamino)-carbonyl]oxime. -
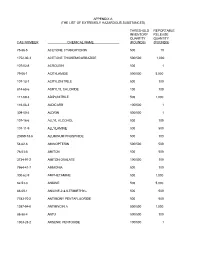
The List of Extremely Hazardous Substances)
APPENDIX A (THE LIST OF EXTREMELY HAZARDOUS SUBSTANCES) THRESHOLD REPORTABLE INVENTORY RELEASE QUANTITY QUANTITY CAS NUMBER CHEMICAL NAME (POUNDS) (POUNDS) 75-86-5 ACETONE CYANOHYDRIN 500 10 1752-30-3 ACETONE THIOSEMICARBAZIDE 500/500 1,000 107-02-8 ACROLEIN 500 1 79-06-1 ACRYLAMIDE 500/500 5,000 107-13-1 ACRYLONITRILE 500 100 814-68-6 ACRYLYL CHLORIDE 100 100 111-69-3 ADIPONITRILE 500 1,000 116-06-3 ALDICARB 100/500 1 309-00-2 ALDRIN 500/500 1 107-18-6 ALLYL ALCOHOL 500 100 107-11-9 ALLYLAMINE 500 500 20859-73-8 ALUMINUM PHOSPHIDE 500 100 54-62-6 AMINOPTERIN 500/500 500 78-53-5 AMITON 500 500 3734-97-2 AMITON OXALATE 100/500 100 7664-41-7 AMMONIA 500 100 300-62-9 AMPHETAMINE 500 1,000 62-53-3 ANILINE 500 5,000 88-05-1 ANILINE,2,4,6-TRIMETHYL- 500 500 7783-70-2 ANTIMONY PENTAFLUORIDE 500 500 1397-94-0 ANTIMYCIN A 500/500 1,000 86-88-4 ANTU 500/500 100 1303-28-2 ARSENIC PENTOXIDE 100/500 1 THRESHOLD REPORTABLE INVENTORY RELEASE QUANTITY QUANTITY CAS NUMBER CHEMICAL NAME (POUNDS) (POUNDS) 1327-53-3 ARSENOUS OXIDE 100/500 1 7784-34-1 ARSENOUS TRICHLORIDE 500 1 7784-42-1 ARSINE 100 100 2642-71-9 AZINPHOS-ETHYL 100/500 100 86-50-0 AZINPHOS-METHYL 10/500 1 98-87-3 BENZAL CHLORIDE 500 5,000 98-16-8 BENZENAMINE, 3-(TRIFLUOROMETHYL)- 500 500 100-14-1 BENZENE, 1-(CHLOROMETHYL)-4-NITRO- 500/500 500 98-05-5 BENZENEARSONIC ACID 10/500 10 3615-21-2 BENZIMIDAZOLE, 4,5-DICHLORO-2-(TRI- 500/500 500 FLUOROMETHYL)- 98-07-7 BENZOTRICHLORIDE 100 10 100-44-7 BENZYL CHLORIDE 500 100 140-29-4 BENZYL CYANIDE 500 500 15271-41-7 BICYCLO[2.2.1]HEPTANE-2-CARBONITRILE,5- -

9-2909-00045/02001 Effective Date: 10/16/2014 Expiration Date: 10/15/2019
Facility DEC ID: 9290900045 PERMIT Under the Environmental Conservation Law (ECL) IDENTIFICATION INFORMATION Permit Type: Air State Facility Permit ID: 9-2909-00045/02001 Effective Date: 10/16/2014 Expiration Date: 10/15/2019 Permit Issued To:TWIN LAKE CHEMICAL INC 520 MILL ST PO BOX 411 LOCKPORT, NY 14094-0411 Contact: JAMES D HODAN TWIN LAKE CHEMICAL INC PO BOX 411 LOCKPORT, NY 14095 (716) 433-3824 Facility: TWIN LAKE CHEMICAL INC 520 MILL ST LOCKPORT, NY 14094 Contact: WILLIAM CASWELL TWIN LAKE CHEMICAL INC 520 MILL ST LOCKPORT, NY 14094-1712 (716) 433-3824 Description: This Air State Facility permit incorporates monitoring conditions for Twin Lake Chemical located in Lockport New York. The facility is a manufacturer of various organic acid chlorides used as intermediaries in the production of other compounds and encompasses two main production buildings and 8 reactors. The primary products produced are trimellitic trichloride, trimellitic Anhydride Monoacid chloride,isophthaloyl chloride, orthophthaloyl chloride, terephthaloyl chloride, and phosphorous pentachloride (2-200 gallon nickel reactors). Phosgene and thionyl chloride are used as chlorinating agents. The phosgene is purchased in 1 ton containers from Vandemark Chemical located next to the facility. Raw materials are added to the batch reactors along with a chlorinating agent and a catalyst. Prior to opening the reactor for chemical addition, the reactor is put under vacuum to remove gases. During the reaction process, the reactor is under slight pressure. Reactor temperatures are monitored. After the reaction is completed, the material is transferred to a distillation unit to refine and separate the product. Air strippers remove chlorinated hydrocarbons from wastewater prior to discharge to Lockport WWTP. -

744 Hydrolysis of Chiral Organophosphorus Compounds By
[Frontiers in Bioscience, Landmark, 26, 744-770, Jan 1, 2021] Hydrolysis of chiral organophosphorus compounds by phosphotriesterases and mammalian paraoxonase-1 Antonio Monroy-Noyola1, Damianys Almenares-Lopez2, Eugenio Vilanova Gisbert3 1Laboratorio de Neuroproteccion, Facultad de Farmacia, Universidad Autonoma del Estado de Morelos, Morelos, Mexico, 2Division de Ciencias Basicas e Ingenierias, Universidad Popular de la Chontalpa, H. Cardenas, Tabasco, Mexico, 3Instituto de Bioingenieria, Universidad Miguel Hernandez, Elche, Alicante, Spain TABLE OF CONTENTS 1. Abstract 2. Introduction 2.1. Organophosphorus compounds (OPs) and their toxicity 2.2. Metabolism and treatment of OP intoxication 2.3. Chiral OPs 3. Stereoselective hydrolysis 3.1. Stereoselective hydrolysis determines the toxicity of chiral compounds 3.2. Hydrolysis of nerve agents by PTEs 3.2.1. Hydrolysis of V-type agents 3.3. PON1, a protein restricted in its ability to hydrolyze chiral OPs 3.4. Toxicity and stereoselective hydrolysis of OPs in animal tissues 3.4.1. The calcium-dependent stereoselective activity of OPs associated with PON1 3.4.2. Stereoselective hydrolysis commercial OPs pesticides by alloforms of PON1 Q192R 3.4.3. PON1, an enzyme that stereoselectively hydrolyzes OP nerve agents 3.4.4. PON1 recombinants and stereoselective hydrolysis of OP nerve agents 3.5. The activity of PTEs in birds 4. Conclusions 5. Acknowledgments 6. References 1. ABSTRACT Some organophosphorus compounds interaction of the racemic OPs with these B- (OPs), which are used in the manufacturing of esterases (AChE and NTE) and such interactions insecticides and nerve agents, are racemic mixtures have been studied in vivo, ex vivo and in vitro, using with at least one chiral center with a phosphorus stereoselective hydrolysis by A-esterases or atom. -

Benzyl Chlorid Final
Survey of benzyl chloride (CAS no. 100-44-7) Part of the LOUS-review [Series Title and year] Consultation draft Title: Editing: Survey of benzyl chloride (CAS no. 100-44-7) Pia Brunn Poulsen, FORCE Technology Maria Strandesen, FORCE Technology Anders Schmidt, FORCE Technology Published by: Photography: The Danish Environmental Protection Agency [Name] Strandgade 29 1401 Copenhagen K Denmark Illustration: www.mst.dk/english [Name] Year: Map: [xxxx] [Name] ISBN no. [xxxxxx] Disclaimer: When the occasion arises, the Danish Environmental Protection Agency will publish reports and papers concerning research and development projects within the environmental sector, financed by study grants provided by the Danish Environmental Protection Agency. It should be noted that such publications do not necessarily reflect the position or opinion of the Danish Environmental Protection Agency. However, publication does indicate that, in the opinion of the Danish Environmental Protection Agency, the content represents an important contribution to the debate surrounding Danish environmental policy. While the information provided in this report is believed to be accurate, The Danish Environmental Protection Agency disclaims any responsibility for possible inaccuracies or omissions and consequences that may flow from them. Neither the Danish Environmental Protection Agency nor FORCE Technology or any individual involved in the preparation of this publication shall be liable for any injury, loss, damage or prejudice of any kind that may be caused by persons who have acted based on their understanding of the information contained in this publication. Sources must be acknowledged. 2 Survey of benzyl chloride (CAS no. 100-44-7) Contents Preface ...................................................................................................................... 6 Summary and conclusions ......................................................................................... 8 Sammenfatning og konklusion ............................................................................... -

The Preparation of Certain Organic Chloroformates and Carbonates
Brigham Young University BYU ScholarsArchive Theses and Dissertations 1947-04-01 The preparation of certain organic chloroformates and carbonates Robert E. Brailsford Brigham Young University - Provo Follow this and additional works at: https://scholarsarchive.byu.edu/etd BYU ScholarsArchive Citation Brailsford, Robert E., "The preparation of certain organic chloroformates and carbonates" (1947). Theses and Dissertations. 8175. https://scholarsarchive.byu.edu/etd/8175 This Thesis is brought to you for free and open access by BYU ScholarsArchive. It has been accepted for inclusion in Theses and Dissertations by an authorized administrator of BYU ScholarsArchive. For more information, please contact [email protected], [email protected]. THE PREPARATI OH OF CERTAI N 0RG.1NIC CHLOROFORJ.'\Ll.TES AND CARBON T Thesis ubmitted to the Department of Chemistry Brigham Young University ~ .. ... "') .~ . ~ . "'). ... .. .. .. .. , ... .. ... : : ....: . ..-. ~ ..·.: : ..: ...• : ·.. ~ . : ,.. .~ : :. : ·: : ··.... ; ~ : ·. : : .: . : . : : : ••• .... •." •,.r_·: -••• ~ .... In Parti a l Fulfillment of the Re~uirements for the Degree Master of cienoe 147143 by Robert E. Brailsford .tipril 1947 This Thesis by Robert E. Brailsford is accepted in its present farm by the Department of Chemistry of Brigham Young University as satisfying the ·rhesis requirement for the degree of Master of Science. PREFACE flhile working for the Hooker Electrochemical Company of Niagara Falls , New York , from April 3 , 1943 , to January 30 , 1946 , the writer became interested in organic chloroformates and. carbonates , an interest instigated by requests from B. F . Goodrich Company for a number of samples . fter returning to Brigham Young University that preliminary interest was revived and the experi - mental work of this thesis was performed. under the direction of Dr . Charles ' . :Maw and Professor Joseph K. -

United States Patent (19) 11 Patent Number: 6,028, 182 Uhlmann Et Al
US006028182A United States Patent (19) 11 Patent Number: 6,028, 182 Uhlmann et al. (45) Date of Patent: *Feb. 22, 2000 54 METHYLPHOSPHONIC ACID ESTERS, Roelen, “Synthesis of alkylphosphon(othio)ate analogues of PROCESSES FOR THEIR PREPARATION, DNA.” Tetrahedron Letters, vol. 33, 1992, pp. 2357–2360. AND THEIR USE Meier, “O-Alkyl-5", 5'-dinucleoside-phosphates as com bined prodrugs of antiviral and antibiotic compounds,” 75 Inventors: Eugen Uhlmann, Glashütten; Chris Bioorganic Med. Chem. Lett., vol. 1, 1991, pp. 527-530. Meier, Bad Homburg, both of Germany Chris Meier et al., Lipophilic C-Hydroxybenzylphospho 73 Assignee: Hoechst Aktiengesellschaft, Frankfurt nates as Prodrugs of 3'-Azido-2, 3-dideoxythymidine am Main, Germany (AZT), Liesbigs Ann. 1995, 2195-2202. Chris Meier et al., Homo * Notice: This patent issued on a continued pros Dinucleoside-C-Hydroxyphoshonate Diesters as Prodrugs ecution application filed under 37 CFR of the Antiviral Nuceloside Analogues 2',3'-Dideoxythy 1.53(d), and is subject to the twenty year midine and 3'-Azido-2',3'-Dideoxythymidine, Nucelosides patent term provisions of 35 U.S.C. & Nucelositeda, 14(3-5), 759–762 (1995). 154(a)(2). Chris Meier, Lipohilic 5', 5-0-Dinucleoside-O-hydroxybenzylphophonic Acid 21 Appl. No.: 08/578,686 Esters as Potential Prodrugs of 2, 3'-Dideoxythymidine 22 PCT Filed: Jun. 29, 1994 (ddT), Agnew. Chemical Int. Ed. Engl 1993, vol. 32, No. 12, pp. 1704-1706. 86 PCT No.: PCT/EP94/02121 Yoshino et al. “Organic phosphorus compounds. 2. Synthe S371 Date: Jan. 2, 1996 sis and coronary vasodilator activity of (benzothiazolylben Zyl)phosphonate derivatives.” J. -

Benzyl Chloroformate (CHLOROFORMIC ACID, BENZYL ESTER)
Rev B Benzyl Chloroformate (CHLOROFORMIC ACID, BENZYL ESTER) C8H7O2Cl Molecular Weight = 170.6 CAS# 501‐53‐1 SPECIFICATIONS Assay: 98.% min. Color (APHA): 50 max. Benzyl Alcohol: 0.1% max. Hydrogen Chloride: 0.1% max. Benzyl Chloride: 1.5% max. Phosgene: 0.1% max. Dibenzyl Carbonate: 0.5% max. Iron: 1.5 PPM max. PHYSICAL PROPERTIES Appearance: Clear liquid free of visible contaminants BP: Decomposes at elevated temperature Odor: Pungent Density: 1.195 ‐1.22 MP/Range: ‐30°C Flash Point: 126°C NOTICE: The technical information and suggestions for use made herein are based on VanDeMark’s research and experience and are believed to be reliable, but such information and suggestions do not constitute a warranty, and no patent liability can be assumed. This publication is not to be taken as a license to operate under or infringe on any patents. Since VanDeMark has no control over the conditions under which the product is transported, stored, handled, used or applied, buyer must determine for himself by preliminary tests or otherwise, the suitability of the product for his purposes. VanDeMark’s liability on any basis is limited to the price of the product used. The information in this bulletin supersedes all previously issued bulletins on the subject matter covered. VanDeMark Benzyl Chloroformate APPLICATIONS SPILLS AND DISPOSAL Benzyl Chloroformate is a reactive chemical intermediate Use personal protective equipment (see MSDS). used in the synthesis of pharmaceutical and agrochemical Evacuate personnel to safe areas. Dike far ahead of products. It is used as a reagent in peptide synthesis to liquid spill for later disposal.