Important Interactions in Peptide-Based Transfection Agents, Sugars As Chiral Scaffolds, and Molecular Imprinting of Nerve Gases
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

( 12 ) United States Patent ( 10 ) Patent No .: US 10,751,310 B2 Freeman Et Al
US010751310B2 ( 12 ) United States Patent ( 10 ) Patent No .: US 10,751,310 B2 Freeman et al . ( 45 ) Date of Patent : Aug. 25 , 2020 ( 54 ) PREVENTION , TREATMENT AND ( 56 ) References Cited REVERSAL OF DISEASE USING THERAPEUTICALLY EFFECTIVE U.S. PATENT DOCUMENTS AMOUNTS OF DICARBOXYLIC ACID 3,527,789 A 9/1970 Payne COMPOUNDS 4,166,913 A 9/1979 Kesling , Jr. et al . 6,528,499 B1 * 3/2003 Kozikowski C07C 59/347 ( 71 ) Applicant: UNIVERSITY OF 514/574 8,324,277 B2 12/2012 Freeman PITTSBURGH — OF THE 8,735,449 B2 5/2014 Freeman COMMONWEALTH SYSTEM OF 9,066,902 B2 6/2015 Freeman et al . HIGHER EDUCATION , Pittsburgh , 9,186,408 B2 11/2015 Freeman et al . PA (US ) 9,700,534 B2 7/2017 Freeman et al . 9,750,725 B2 9/2017 Freeman et al . 10,213,417 B2 2/2019 Freeman et al . ( 72 ) Inventors : Bruce A. Freeman , Pittsburgh , PA 10,258,589 B2 4/2019 Freeman et al . ( US ) ; Francisco J. Schopfer , 2015/0018417 Al 1/2015 Freeman et al . Pittsburgh , PA ( US ) FOREIGN PATENT DOCUMENTS ( 73 ) Assignee : University of Pittsburgh — of the CN 103705499 4/2014 Commonwealth System of Higher DE 102011118462 5/2013 Education , Pittsburgh , PA ( US ) GB 1153464 5/1969 WO WO 2002/022627 3/2002 WO WO 2009/017802 2/2009 ( * ) Notice : Subject to any disclaimer , the term of this WO WO 2009/112455 9/2009 patent is extended or adjusted under 35 WO WO 2010/005521 1/2010 U.S.C. 154 ( b ) by 0 days . WO WO 2010/014889 2/2010 WO WO 2011/014261 2/2011 WO WO 2013/116753 8/2013 ( 21 ) Appl. -

Production of Butanol by Clostridium Acetobutyucum in Extractive Fermentation System
PRODUCTION OF BUTANOL BY CLOSTRIDIUM ACETOBUTYUCUM IN EXTRACTIVE FERMENTATION SYSTEM Shigeo ISHII, Masahito TAYA and Takeshi KOBAYASHI Department of Chemical Engineering, Faculty of Engineering, Nagoya University, Nagoya 464 Key Words: Biochemical Engineering, Extractive Fermentation, Butanol Production, Aliphatic Alcohol, Clostridium acetobutylicum Anextractive fermentation system was developed to prevent end-product inhibition of Clostridium aceto- butylicum IAM 19012, which mainly produces butanol and acetone. Butanol exhibited greater toxicity to the microorganism than acetone, and its growth was completely inhibited above 10 kg/m3 of butanol. As an extracting solvent suitable for acetone-butanol fermentation, oleyl alcohol (cw-9-octadecen-l-ol) and C-20 guerbet alcohol (branched-chain alcohol of carbon number, 20) were selected from among29 organic compounds, based on their nontoxicity to the microorganism. These two solvents had high partition coefficients for butanol, and could be reused without deterioration. In fermentation with the solvent (solvent phase : aqueous phase=2 : 5 (v/v)), the viability of the microorganism was resumed by the liquid-liquid extraction of butanol from the broth, and the amount of butanol produced was 2.6 times that in fermentation without extraction. tation by liquid-liquid extraction. To date, a few Intr oduction studies on extractive fermentation have been carried Microbial production of acetone and butanol is a out for ethanol fermentation by yeast.5'10'18) traditional fermentation process. After World War II, The aim of the present study is to develop a new however, the fermentation process for acetone- strategy which combines both physical liquid-liquid butanol production was superseded by chemical extraction and biological fermentation processes, and synthetic processes using petroleum-based feed- furthermore to achieve improved production of bu- stocks, except for a fermentation plant in South Africa.15) tanol and acetone by extractive fermentation. -

Chemical Name Federal P Code CAS Registry Number Acutely
Acutely / Extremely Hazardous Waste List Federal P CAS Registry Acutely / Extremely Chemical Name Code Number Hazardous 4,7-Methano-1H-indene, 1,4,5,6,7,8,8-heptachloro-3a,4,7,7a-tetrahydro- P059 76-44-8 Acutely Hazardous 6,9-Methano-2,4,3-benzodioxathiepin, 6,7,8,9,10,10- hexachloro-1,5,5a,6,9,9a-hexahydro-, 3-oxide P050 115-29-7 Acutely Hazardous Methanimidamide, N,N-dimethyl-N'-[2-methyl-4-[[(methylamino)carbonyl]oxy]phenyl]- P197 17702-57-7 Acutely Hazardous 1-(o-Chlorophenyl)thiourea P026 5344-82-1 Acutely Hazardous 1-(o-Chlorophenyl)thiourea 5344-82-1 Extremely Hazardous 1,1,1-Trichloro-2, -bis(p-methoxyphenyl)ethane Extremely Hazardous 1,1a,2,2,3,3a,4,5,5,5a,5b,6-Dodecachlorooctahydro-1,3,4-metheno-1H-cyclobuta (cd) pentalene, Dechlorane Extremely Hazardous 1,1a,3,3a,4,5,5,5a,5b,6-Decachloro--octahydro-1,2,4-metheno-2H-cyclobuta (cd) pentalen-2- one, chlorecone Extremely Hazardous 1,1-Dimethylhydrazine 57-14-7 Extremely Hazardous 1,2,3,4,10,10-Hexachloro-6,7-epoxy-1,4,4,4a,5,6,7,8,8a-octahydro-1,4-endo-endo-5,8- dimethanonaph-thalene Extremely Hazardous 1,2,3-Propanetriol, trinitrate P081 55-63-0 Acutely Hazardous 1,2,3-Propanetriol, trinitrate 55-63-0 Extremely Hazardous 1,2,4,5,6,7,8,8-Octachloro-4,7-methano-3a,4,7,7a-tetra- hydro- indane Extremely Hazardous 1,2-Benzenediol, 4-[1-hydroxy-2-(methylamino)ethyl]- 51-43-4 Extremely Hazardous 1,2-Benzenediol, 4-[1-hydroxy-2-(methylamino)ethyl]-, P042 51-43-4 Acutely Hazardous 1,2-Dibromo-3-chloropropane 96-12-8 Extremely Hazardous 1,2-Propylenimine P067 75-55-8 Acutely Hazardous 1,2-Propylenimine 75-55-8 Extremely Hazardous 1,3,4,5,6,7,8,8-Octachloro-1,3,3a,4,7,7a-hexahydro-4,7-methanoisobenzofuran Extremely Hazardous 1,3-Dithiolane-2-carboxaldehyde, 2,4-dimethyl-, O- [(methylamino)-carbonyl]oxime 26419-73-8 Extremely Hazardous 1,3-Dithiolane-2-carboxaldehyde, 2,4-dimethyl-, O- [(methylamino)-carbonyl]oxime. -

Listed Below Are All of the Revised Titles in the Description and Solubility Reference Table of USP 36-NF 31 Second Supplement
Listed below are all of the revised titles in the Description and Solubility reference table of USP 36-NF 31 Second Supplement. For additional information please see general information chapter Excipient Performance <1059> and USP and NF Excipients, Listed by Functional Category, published in the USP 36- NF 31 Second Supplement. Acacia Carbomer Interpolymer Acetic Acid Carbon Dioxide Glacial Acetic Acid Carboxymethylcellulose Calcium Adipic Acid Carboxymethylcellulose Sodium Agar Enzymatically-Hydrolyzed Alcohol Carboxymethylcellulose Sodium Alfadex Carmellose Alginic Acid Carrageenan Almond Oil Castor Oil Aluminum Monostearate Hydrogenated Castor Oil Aluminum Oxide Cellaburate Amino Methacrylate Copolymer Cellacefate Aminobenzoic Acid Cellulose Acetate Strong Ammonia Solution Microcrystalline Cellulose Ammonio Methacrylate Copolymer Silicified Microcrystalline Cellulose Ammonio Methacrylate Copolymer Dispersion Powdered Cellulose Ammonium Carbonate Cetostearyl Alcohol Ammonium Chloride Cetyl Alcohol Ammonium Phosphate Cetyl Esters Wax Anethole Cetylpyridinium Chloride Behenoyl Polyoxylglycerides Chlorobutanol Benzaldehyde Chloroxylenol Benzalkonium Chloride Cholesterol Benzyl Alcohol Anhydrous Citric Acid Benzyl Benzoate Citric Acid Monohydrate Betadex Coconut Oil Betadex Sulfobutyl Ether Sodium Hydrogenated Coconut Oil Boric Acid Copovidone Butane Corn Syrup Calcium Acetate Corn Syrup Solids Calcium Carbonate Creatinine Calcium Chloride Croscarmellose Sodium Calcium Hydroxide Crospovidone Calcium Lactate Denatonium Benzoate Dibasic -

Official Journal of the European Communities on the Hygiene Of
No L 21 /42 EN Official Journal of the European Communities 27 . 1 . 96 COMMISSION DIRECTIVE 96/3/EC of 26 January 1 996 granting a derogation from certain provisions of Council Directive 93/43/EEC on the hygiene of foodstuffs as regards the transport of bulk liquid oils and fats by sea (Text with EEA relevance) THE COMMISSION OF THE EUROPEAN COMMUNITIES, whereas the measures provided for in this Directive are in compliance with the opinion of the Standing Having regard to the Treaty establishing the European Committee for Foodstuffs, Community, Having regard to Council Directive 93/43/EEC of 14 June 1993 on the hygiene of foodstuffs ('), and in parti HAS ADOPTED THIS DIRECTIVE : cular Article 3 (3) thereof, Whereas information shows that the application of the second subparagraph of paragraph 2 of Chapter IV of the Article 1 Annex to Directive 93/43/EEC relating to the transport of bulk foodstuffs in liquid, granulate or powdered form in This Directive derogates from the second subparagraph of receptacles and/or containers/tankers reserved for the paragraph 2 of Chapter IV of the Annex to Directive transport of foodstuffs, is not practical and imposes an 93/43/EEC and lays down equivalent conditions to ensure unduly onerous burden on food business when applied to the protection of public health and the safety and whole the transport in sea-going vessels of liquid oils and fats someness of the foodstuffs concerned . intended for, or likely to be used for, human consump tion ; Article 2 Whereas, however, it is necessary to ensure that the granting of a derogation provides equivalent protection to public health, by attaching conditions to the terms of 1 . -
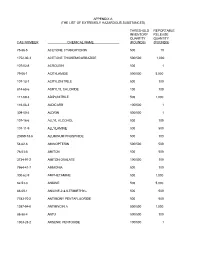
The List of Extremely Hazardous Substances)
APPENDIX A (THE LIST OF EXTREMELY HAZARDOUS SUBSTANCES) THRESHOLD REPORTABLE INVENTORY RELEASE QUANTITY QUANTITY CAS NUMBER CHEMICAL NAME (POUNDS) (POUNDS) 75-86-5 ACETONE CYANOHYDRIN 500 10 1752-30-3 ACETONE THIOSEMICARBAZIDE 500/500 1,000 107-02-8 ACROLEIN 500 1 79-06-1 ACRYLAMIDE 500/500 5,000 107-13-1 ACRYLONITRILE 500 100 814-68-6 ACRYLYL CHLORIDE 100 100 111-69-3 ADIPONITRILE 500 1,000 116-06-3 ALDICARB 100/500 1 309-00-2 ALDRIN 500/500 1 107-18-6 ALLYL ALCOHOL 500 100 107-11-9 ALLYLAMINE 500 500 20859-73-8 ALUMINUM PHOSPHIDE 500 100 54-62-6 AMINOPTERIN 500/500 500 78-53-5 AMITON 500 500 3734-97-2 AMITON OXALATE 100/500 100 7664-41-7 AMMONIA 500 100 300-62-9 AMPHETAMINE 500 1,000 62-53-3 ANILINE 500 5,000 88-05-1 ANILINE,2,4,6-TRIMETHYL- 500 500 7783-70-2 ANTIMONY PENTAFLUORIDE 500 500 1397-94-0 ANTIMYCIN A 500/500 1,000 86-88-4 ANTU 500/500 100 1303-28-2 ARSENIC PENTOXIDE 100/500 1 THRESHOLD REPORTABLE INVENTORY RELEASE QUANTITY QUANTITY CAS NUMBER CHEMICAL NAME (POUNDS) (POUNDS) 1327-53-3 ARSENOUS OXIDE 100/500 1 7784-34-1 ARSENOUS TRICHLORIDE 500 1 7784-42-1 ARSINE 100 100 2642-71-9 AZINPHOS-ETHYL 100/500 100 86-50-0 AZINPHOS-METHYL 10/500 1 98-87-3 BENZAL CHLORIDE 500 5,000 98-16-8 BENZENAMINE, 3-(TRIFLUOROMETHYL)- 500 500 100-14-1 BENZENE, 1-(CHLOROMETHYL)-4-NITRO- 500/500 500 98-05-5 BENZENEARSONIC ACID 10/500 10 3615-21-2 BENZIMIDAZOLE, 4,5-DICHLORO-2-(TRI- 500/500 500 FLUOROMETHYL)- 98-07-7 BENZOTRICHLORIDE 100 10 100-44-7 BENZYL CHLORIDE 500 100 140-29-4 BENZYL CYANIDE 500 500 15271-41-7 BICYCLO[2.2.1]HEPTANE-2-CARBONITRILE,5- -

Hexanol Biosynthesis from Syngas by Clostridium Carboxidivorans P7 Is Enhanced by In‑Line Extraction with a Biocompatible Solvent to Avoid Product Toxicity
Hexanol biosynthesis from syngas by Clostridium carboxidivorans P7 is enhanced by in‐line extraction with a biocompatible solvent to avoid product toxicity Patrick Kottenhahn Fraunhofer-Institut fur Molekularbiologie und Angewandte Oekologie Gabriele Philipps Fraunhofer-Institut fur Molekularbiologie und Angewandte Oekologie Stefan Jennewein ( [email protected] ) Fraunhofer-Institut fur Molekularbiologie und Angewandte Oekologie https://orcid.org/0000-0002- 1625-5484 Research Keywords: oleyl alcohol, product toxicity, bioeconomy, carbon xation, C1 compounds, Clostridium ljungdahlii, in situ extraction, biofuels Posted Date: February 18th, 2020 DOI: https://doi.org/10.21203/rs.2.23824/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 1/17 Abstract Background Clostridium carboxidivorans P7 has the rare ability to metabolize syngas – a mixture of H2, CO and CO2 – by converting it directly into industrially relevant alcohols (hexanol, butanol and ethanol) and the corresponding acids (caproate, butyrate and acetate). The product titers and ratios are highly dependent on the fermentation parameters and the composition of the syngas and growth medium. The hexanol titers produced by C. carboxidivorans P7 have recently been improved by optimizing these conditions, but little is known about the toxicity of hexanol towards Clostridium species. We hypothesized that the hexanol titers currently produced by C. carboxidivorans P7 are limited by product toxicity. Results We tested our hypothesis by exposing C. carboxidivorans P7 to different concentrations of hexanol and found that growth inhibition started at 10–12 mM, with an IC50 of 17.5 ± 1.6 mM. The presence of 20 mM hexanol was acutely toxic to C. -

Benzyl Chlorid Final
Survey of benzyl chloride (CAS no. 100-44-7) Part of the LOUS-review [Series Title and year] Consultation draft Title: Editing: Survey of benzyl chloride (CAS no. 100-44-7) Pia Brunn Poulsen, FORCE Technology Maria Strandesen, FORCE Technology Anders Schmidt, FORCE Technology Published by: Photography: The Danish Environmental Protection Agency [Name] Strandgade 29 1401 Copenhagen K Denmark Illustration: www.mst.dk/english [Name] Year: Map: [xxxx] [Name] ISBN no. [xxxxxx] Disclaimer: When the occasion arises, the Danish Environmental Protection Agency will publish reports and papers concerning research and development projects within the environmental sector, financed by study grants provided by the Danish Environmental Protection Agency. It should be noted that such publications do not necessarily reflect the position or opinion of the Danish Environmental Protection Agency. However, publication does indicate that, in the opinion of the Danish Environmental Protection Agency, the content represents an important contribution to the debate surrounding Danish environmental policy. While the information provided in this report is believed to be accurate, The Danish Environmental Protection Agency disclaims any responsibility for possible inaccuracies or omissions and consequences that may flow from them. Neither the Danish Environmental Protection Agency nor FORCE Technology or any individual involved in the preparation of this publication shall be liable for any injury, loss, damage or prejudice of any kind that may be caused by persons who have acted based on their understanding of the information contained in this publication. Sources must be acknowledged. 2 Survey of benzyl chloride (CAS no. 100-44-7) Contents Preface ...................................................................................................................... 6 Summary and conclusions ......................................................................................... 8 Sammenfatning og konklusion ............................................................................... -

The Preparation of Certain Organic Chloroformates and Carbonates
Brigham Young University BYU ScholarsArchive Theses and Dissertations 1947-04-01 The preparation of certain organic chloroformates and carbonates Robert E. Brailsford Brigham Young University - Provo Follow this and additional works at: https://scholarsarchive.byu.edu/etd BYU ScholarsArchive Citation Brailsford, Robert E., "The preparation of certain organic chloroformates and carbonates" (1947). Theses and Dissertations. 8175. https://scholarsarchive.byu.edu/etd/8175 This Thesis is brought to you for free and open access by BYU ScholarsArchive. It has been accepted for inclusion in Theses and Dissertations by an authorized administrator of BYU ScholarsArchive. For more information, please contact [email protected], [email protected]. THE PREPARATI OH OF CERTAI N 0RG.1NIC CHLOROFORJ.'\Ll.TES AND CARBON T Thesis ubmitted to the Department of Chemistry Brigham Young University ~ .. ... "') .~ . ~ . "'). ... .. .. .. .. , ... .. ... : : ....: . ..-. ~ ..·.: : ..: ...• : ·.. ~ . : ,.. .~ : :. : ·: : ··.... ; ~ : ·. : : .: . : . : : : ••• .... •." •,.r_·: -••• ~ .... In Parti a l Fulfillment of the Re~uirements for the Degree Master of cienoe 147143 by Robert E. Brailsford .tipril 1947 This Thesis by Robert E. Brailsford is accepted in its present farm by the Department of Chemistry of Brigham Young University as satisfying the ·rhesis requirement for the degree of Master of Science. PREFACE flhile working for the Hooker Electrochemical Company of Niagara Falls , New York , from April 3 , 1943 , to January 30 , 1946 , the writer became interested in organic chloroformates and. carbonates , an interest instigated by requests from B. F . Goodrich Company for a number of samples . fter returning to Brigham Young University that preliminary interest was revived and the experi - mental work of this thesis was performed. under the direction of Dr . Charles ' . :Maw and Professor Joseph K. -

Benzyl Chloroformate (CHLOROFORMIC ACID, BENZYL ESTER)
Rev B Benzyl Chloroformate (CHLOROFORMIC ACID, BENZYL ESTER) C8H7O2Cl Molecular Weight = 170.6 CAS# 501‐53‐1 SPECIFICATIONS Assay: 98.% min. Color (APHA): 50 max. Benzyl Alcohol: 0.1% max. Hydrogen Chloride: 0.1% max. Benzyl Chloride: 1.5% max. Phosgene: 0.1% max. Dibenzyl Carbonate: 0.5% max. Iron: 1.5 PPM max. PHYSICAL PROPERTIES Appearance: Clear liquid free of visible contaminants BP: Decomposes at elevated temperature Odor: Pungent Density: 1.195 ‐1.22 MP/Range: ‐30°C Flash Point: 126°C NOTICE: The technical information and suggestions for use made herein are based on VanDeMark’s research and experience and are believed to be reliable, but such information and suggestions do not constitute a warranty, and no patent liability can be assumed. This publication is not to be taken as a license to operate under or infringe on any patents. Since VanDeMark has no control over the conditions under which the product is transported, stored, handled, used or applied, buyer must determine for himself by preliminary tests or otherwise, the suitability of the product for his purposes. VanDeMark’s liability on any basis is limited to the price of the product used. The information in this bulletin supersedes all previously issued bulletins on the subject matter covered. VanDeMark Benzyl Chloroformate APPLICATIONS SPILLS AND DISPOSAL Benzyl Chloroformate is a reactive chemical intermediate Use personal protective equipment (see MSDS). used in the synthesis of pharmaceutical and agrochemical Evacuate personnel to safe areas. Dike far ahead of products. It is used as a reagent in peptide synthesis to liquid spill for later disposal. -

Benzyl Chloride
Right to Know Hazardous Substance Fact Sheet Common Name: BENZYL CHLORIDE Synonyms: Chloromethyl Benzene; alpha-Chlorotoluene CAS Number: 100-44-7 Chemical Name: Benzene, (Chloromethyl)- RTK Substance Number: 0217 Date: July 2002 Revision: November 2010 DOT Number: UN 1738 Description and Use EMERGENCY RESPONDERS >>>> SEE LAST PAGE Benzyl Chloride is a colorless liquid with a strong, irritating Hazard Summary odor that causes tearing of the eyes. It is used in making dyes, Hazard Rating NJDOH NFPA plasticizers, drugs, lubricants, resins and cosmetics. HEALTH - 3 FLAMMABILITY - 2 f ODOR THRESHOLD = 0.041 ppm REACTIVITY - 1 f Odor thresholds vary greatly. Do not rely on odor alone to determine potentially hazardous exposures. CARCINOGEN CORROSIVE POISONOUS GASES ARE PRODUCED IN FIRE CONTAINERS MAY EXPLODE IN FIRE Reasons for Citation Hazard Rating Key: 0=minimal; 1=slight; 2=moderate; 3=serious; 4=severe f Benzyl Chloride is on the Right to Know Hazardous Substance List because it is cited by OSHA, ACGIH, DOT, f Benzyl Chloride can affect you when inhaled and may be NIOSH, DEP, IARC, IRIS, NFPA and EPA. absorbed through the skin. f This chemical is on the Special Health Hazard Substance f Benzyl Chloride is a CARCINOGEN and MUTAGEN. List. HANDLE WITH EXTREME CAUTION. f Benzyl Chloride is a CORROSIVE CHEMICAL and contact can severely irritate and burn the skin and eyes with possible eye damage. SEE GLOSSARY ON PAGE 5. f Inhaling Benzyl Chloride can irritate the nose and throat. f Inhaling Benzyl Chloride can irritate the lungs. Higher FIRST AID exposures may cause a build-up of fluid in the lungs Eye Contact (pulmonary edema), a medical emergency. -

OLEYL ALCOHOL Alcohol Oleicus OLIVE LEAF Oleae Folium
Oleyl alcohol EUROPEAN PHARMACOPOEIA 5.0 — arachidic acid: not more than 2.0 per cent, Composition of fatty alcohols. Gas chromatography — eicosenoic acid:notmorethan2.0percent. (2.2.28): use the normalisation procedure. Ethylene oxide and dioxan (2.4.25). Not more than 1 ppm Test solution. Mix 25 mg of the substance to be examined of ethylene oxide and 10 ppm of dioxan. with 1.0 ml of methylene chloride R. Heavy metals (2.4.8). 2.0 g complies with limit test C for Reference solution (a). Dissolve 25 mg of each of arachidyl heavy metals (10 ppm). Prepare the standard using 2 ml of alcohol R, linolenyl alcohol R, linoleyl alcohol R, oleyl lead standard solution (10 ppm Pb) R. alcohol R, palmityl alcohol R and stearyl alcohol R in methylene chloride R and dilute to 5 ml with the same Water (2.5.12). Not more than 1.0 per cent, determined solvent. Dilute 1 ml of this solution to 5 ml with methylene on 1.0 g by the semi-micro determination of water. Use chloride R. amixtureof30volumesofanhydrous methanol R and Reference solution (b). Dissolve 10 mg of linoleyl alcohol R 70 volumes of methylene chloride R as solvent. and 1 g of oleyl alcohol R in methylenechlorideRand Total ash (2.4.16). Not more than 0.1 per cent, determined dilute to 40 ml with the same solvent. on 1.0 g. Column: STORAGE — size: l =30m,Ø=0.32mm, Store protected from light and at room temperature. — stationary phase: poly(dimethyl)siloxane R (film thickness 1 µm).