An Integrated Pathway for Building Regional Phylogenies For
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Fish and Invertebrate Bycatch and Discards in New Zealand Hoki, Hake, and Ling Fisheries from 1990–91 Until 2012–13
Fish and invertebrate bycatch and discards in New Zealand hoki, hake, or ling trawl fisheries from 1990–91 until 2012–13 New Zealand Aquatic Environment and Biodiversity Report No. 163 S.L. Ballara R.L. O’Driscoll ISSN 1179-6480 (online) ISBN 978-1-77665-111-5 (online) November 2015 Requests for further copies should be directed to: Publications Logistics Officer Ministry for Primary Industries PO Box 2526 WELLINGTON 6140 Email: [email protected] Telephone: 0800 00 83 33 Facsimile: 04-894 0300 This publication is also available on the Ministry for Primary Industries websites at: http://www.mpi.govt.nz/news-resources/publications.aspx http://fs.fish.govt.nz go to Document library/Research reports © Crown Copyright - Ministry for Primary Industries Table of Contents EXECUTIVE SUMMARY 1 1. INTRODUCTION 3 2. METHODS 5 2.1 Definition of terms 5 2.2 Observer data 5 2.2.1 Data preparation and grooming 6 2.3 Commercial fishing return data 8 2.4 Analysis of factors influencing bycatch and discards 9 2.5 Calculation of bycatch and discard rates 9 2.6 Analysis of temporal trends in bycatch and discards 10 2.7 Comparison of trends in bycatch with data from trawl surveys 11 2.8 Discard information from Catch Landing Returns 11 2.9 Observer-authorised discarding 12 3. RESULTS 12 3.1 Distribution and representativeness of observer data 12 3.2 Comparison of estimators 13 3.3 Bycatch data (excluding discards) 14 3.3.1 Overview of raw bycatch data 14 3.3.2 Regression modelling and stratification of bycatch data 15 3.4 Discard data 15 3.4.1 Overview of -

A New Congrid Eel (Teleostei: Anguilliformes: Congridae) from the Western Pacific, with an Analysis of Its Relationships
Zootaxa 4845 (2): 191–210 ISSN 1175-5326 (print edition) https://www.mapress.com/j/zt/ Article ZOOTAXA Copyright © 2020 Magnolia Press ISSN 1175-5334 (online edition) https://doi.org/10.11646/zootaxa.4845.2.2 http://zoobank.org/urn:lsid:zoobank.org:pub:B2DA6D79-874E-48C1-B054-FEC08546223C A new congrid eel (Teleostei: Anguilliformes: Congridae) from the Western Pacific, with an analysis of its relationships DAVID G. SMITH1*, EMMA S. KARMOVSKAYA2 & JOÃO PAULO CAPRETZ BATISTA DA SILVA3 1Smithsonian Institution, Museum Support Center, MRC-534, 4210 Silver Hill Road, Suitland, MD 20746 [email protected]; https://orcid.org/0000-0002-6354-2427 2Shirshov Institute of Oceanology, Russian Academy of Sciences, Moscow, 117218, Russia [email protected]; https://orcid.org/0000-0002-0636-4265 3Departamento de Sistemática e Ecologia, Centro de Ciências Exatas e da Natureza, Universidade Federal da Paraíba, Castelo Branco, 58051-900, João Pessoa, PB, Brazil [email protected]; https://orcid.org/0000-0002-2373-3421 *Corresponding author Abstract A new species of congrid eel, Bathycongrus villosus sp. nov., is described from the Philippines and Vanuatu. It is similar to some of the small-toothed species currently placed in Bathycongrus and to the species of Bassanago. In this paper we compare the new species to Bassanago albescens (Barnard, 1923) and to Bathycongrus parviporus Karmovskaya, 2011, which it most closely resembles. An analysis of 19 characters shows that it agrees with Bat. parviporus in 16 characters and with Bas. albescens in one. In two characters, the three species are all different. We therefore place it in Bathycongrus. Key words: Taxonomy, Pisces, Bathycongrus, new species Introduction The species described here was discovered independently by two of the authors. -

Bassanago Albescens (Barnard, 1923) En El Atlántico Sudoccidental (35º's-45º's)
Ecología trófica del congrio de profundidad Bassanago albescens (Barnard, 1923) en el Atlántico Sudoccidental (35º'S-45º'S) Item Type Theses and Dissertations Authors Izzo, L.P. Publisher UNMdP Download date 26/09/2021 09:19:12 Item License http://creativecommons.org/licenses/by-nc/3.0/ Link to Item http://hdl.handle.net/1834/5157 Autorización para Publicar en un E-Repositorio de Acceso Abierto Apellido y nombres: IZZO, Luciano P. DNI: Correo electrónico: [email protected] AUTORIZO por la presente a la Biblioteca y Servicio de Documentación INIDEP a publicar en texto completo el trabajo final de Tesis/Monografía/Informe de mi autoría que se detalla, permitiendo la consulta de la misma por Internet, así como la entrega por Biblioteca de copias unitarios a los usuarios que lo soliciten con fines de investigación y estudio. Título del trabajo: "Ecología trófica del congrio de profundidad Bassanago albescens(Barnard, 1923) en el Atlántico 5udoccidental(35°5 - 45°5)" ,40 p. Año: 2010 Título y/o grado que opta: Tesis (licenciatura) Facultad: Universidad Nacional de Mar del Plata, Facultad de Ciencias Exactas y Naturales. Firma: Fecha: ~/7~1, ' / , I .' ASFAAN: OceanDocs: http://hdl.handle.net/1834/ Universidad Nacional de Mar del Plata Facultad de Ciencias Exactas y Naturales Departamento de Ciencias Marinas TESIS DE GRADO LICENCIATURA EN CIENCIAS BIOLÓGICAS Ecología trófica del congrio de profundidad Bassanago albescens (Barnard, 1923) en el Atlántico Sudoccidental (35°S - 45°S). Luciano P. Izzo Mar del Plata – Argentina 2010 Director: -

Hyperoglyphe Antarctica) Fisheries Information Relating to the South Pacific Regional Fisheries Management Organisation
Information describing bluenose (Hyperoglyphe antarctica) fisheries information relating to the South Pacific Regional Fisheries Management Organisation WORKING DRAFT 22 June 2007 1 Overview...........................................................................................................................2 2 Taxonomy.........................................................................................................................3 2.1 Phylum......................................................................................................................3 2.2 Class..........................................................................................................................3 2.3 Order.........................................................................................................................3 2.4 Family.......................................................................................................................3 2.5 Genus and species......................................................................................................3 2.6 Scientific synonyms...................................................................................................3 2.7 Common names.........................................................................................................3 2.8 Molecular (DNA or biochemical) bar coding..............................................................3 3 Species Characteristics.....................................................................................................4 -

DNA Barcoding Identifies Argentine Fishes from Marine and Brackish Waters
DNA Barcoding Identifies Argentine Fishes from Marine and Brackish Waters Ezequiel Mabragan˜ a1,2*, Juan Martı´nDı´az de Astarloa1,2, Robert Hanner3, Junbin Zhang4, Mariano Gonza´lez Castro1,2 1 Laboratorio de Biotaxonomı´a Morfolo´gica y Molecular de Peces, Instituto de Investigaciones Marinas y Costeras, Facultad de Ciencias Exactas y Naturales, Universidad Nacional de Mar del Plata, Mar del Plata, Argentina, 2 Consejo Nacional de Investigaciones Cientı´ficas y Te´cnicas, Argentina, 3 Biodiversity Institute of Ontario and Department of Integrative Biology, University of Guelph, Ontario Canada, 4 College of Fisheries and Life Science, Shanghai Ocean University, Shanghai Abstract Background: DNA barcoding has been advanced as a promising tool to aid species identification and discovery through the use of short, standardized gene targets. Despite extensive taxonomic studies, for a variety of reasons the identification of fishes can be problematic, even for experts. DNA barcoding is proving to be a useful tool in this context. However, its broad application is impeded by the need to construct a comprehensive reference sequence library for all fish species. Here, we make a regional contribution to this grand challenge by calibrating the species discrimination efficiency of barcoding among 125 Argentine fish species, representing nearly one third of the known fauna, and examine the utility of these data to address several key taxonomic uncertainties pertaining to species in this region. Methodology/Principal Findings: Specimens were collected and morphologically identified during crusies conducted between 2005 and 2008. The standard BARCODE fragment of COI was amplified and bi-directionally sequenced from 577 specimens (mean of 5 specimens/species), and all specimens and sequence data were archived and interrogated using analytical tools available on the Barcode of Life Data System (BOLD; www.barcodinglife.org). -

FAR 2018/39 – Trawl Survey of Hoki and Middle-Depth Speciesin the Southland and Sub-Antarctic Areas, November–December 2016
Trawl survey of hoki and middle-depth species in the Southland and Sub-Antarctic areas, November–December 2016 (TAN1614) New Zealand Fisheries Assessment Report 2018/39 R. L. O’Driscoll S.L. Ballara D.J. MacGibbon A.C.G. Schimel ISSN 1179-5352 (online) ISBN 978-1-98-857113-3 (online) November 2018 Requests for further copies should be directed to: Publications Logistics Officer Ministry for Primary Industries PO Box 2526 WELLINGTON 6140 Email: [email protected] Telephone: 0800 00 83 33 Facsimile: 04-894 0300 This publication is also available on the Ministry for Primary Industries websites http://www.mpi.govt.nz/news-and-resources/publications http://fs.fish.govt.nz go to Document library/Research reports © Crown Copyright – Fisheries New Zealand TABLE OF CONTENTS EXECUTIVE SUMMARY ........................................................................................................... 1 1. INTRODUCTION ........................................................................................................... 2 1.1 Project objectives ............................................................................................. 3 2. METHODS .................................................................................................................... 3 2.1 Survey design .................................................................................................. 3 2.2 Vessel and equipment ..................................................................................... 4 2.3 Trawling procedure and biological sampling ................................................... -
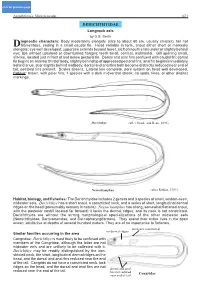
DERICHTHYIDAE Longneck Eels by D.G
click for previous page Anguilliformes: Muraenesocidae 1671 DERICHTHYIDAE Longneck eels by D.G. Smith iagnostic characters: Body moderately elongate (size to about 60 cm, usually smaller), tail not Dfilamentous, ending in a small caudal fin. Head variable in form, snout either short or markedly elongate; eye well developed; upper jaw extends beyond lower, cleft of mouth ends under or slightly behind eye; lips without upturned or downturned flanges; teeth small, conical, multiserial. Gill opening small, slit-like, located just in front of and below pectoral fin. Dorsal and anal fins confluent with caudal fin; dorsal fin begins on anterior third of body, slightly behind tip of appressed pectoral fins; anal fin begins immediately behind anus, at or slightly behind midbody; dorsal and anal fins both become distinctly reduced near end of tail; pectoral fins present. Scales absent. Lateral line complete, pore system on head well developed. Colour: brown, with paler fins; 1 species with a dark midventral streak; no spots, lines, or other distinct markings. Derichthys (after Goode and Bean, 1896) Nessorhamphus (after Robins, 1989) Habitat, biology, and fisheries: The Derichthyidae includes 2 genera and 3 species of small, seldom-seen, midwater eels. Derichthys has a short snout, a constricted neck, and a series of short, longitudinal dermal ridges on the head (presumably sensory in nature). Nessorhamphus has a long, somewhat flattened snout, with the posterior nostril located far forward; it lacks the dermal ridges, and its neck is not constricted. Derichthyids are without the strong morphological specializations of the other midwater eels (Nemichthyidae, Serrivomeridae, and Saccopharyngiformes). They spend their entire lives in the open ocean; adults live at depths of several hundred meters. -

Larvae in the Sargasso Sea: a Molecular Approach Hanna Alfredsson
Prey Selection of European Eel ( Anguilla anguilla) Larvae in the Sargasso Sea: a Molecular Approach Hanna Alfredsson Master Thesis in Aquatic Ecology No: 2009:MBi1 University of Kalmar School of Pure & Applied Natural Sciences Degree project works made at the University of Kalmar, School of Pure and Applied Natural Sciences, can be ordered from: www.hik.se/student or University of Kalmar School of Pure and Applied Natural Sciences SE-391 82 KALMAR SWEDEN Phone + 46 480-44 62 00 Fax + 46 480-44 73 05 e-mail: [email protected] This is a degree project work and the student is responsible for the results and discussions in the report. 2 Prey Selection of European Eel ( Anguilla anguilla ) Larvae in the Sargasso Sea: a Molecular Approach Hanna Alfredsson Master of Science in Aquatic Ecology 120 hp Master Thesis, Aquatic Ecology : 60 hp for Master of Science Supervisor: Associate Professor Lasse Riemann, University of Kalmar Examiner: Associate Professor Catherine Legrand, University of Kalmar Abstract The European eel ( Anguilla anguilla ) migrates to the Sargasso Sea to spawn. Even though the biology of A. anguilla leptocephali in the Sargasso Sea has been studied for several decades, information regarding their diet has remained unknown until now. Previous dietary studies concerning other species of leptocephali in the Pacific Ocean have been limited to the recognition of identifiable prey remains amongst gut contents. Hence, in this study a molecular approach relying on the detection of prey DNA amongst gut contents was used to study dietary profiles of A. anguilla leptocephali in the Sargasso Sea. Leptocephali were collected during the circumglobal Galathea 3 expedition in spring 2007 to the Sargasso Sea. -

Carapace Length-Body Weight Relationship and Condition Factor of Painted Rock Lobster Panulirus Versicolor in Sorong Waters, West Papua, Indonesia 1Yuni M
Carapace length-body weight relationship and condition factor of painted rock lobster Panulirus versicolor in Sorong waters, West Papua, Indonesia 1Yuni M. L. Situmorang, 2Sharifuddin B. A. Omar, 2Joeharnani Tresnati 1 Master Program of Fishery Science, Postgraduate School, Hasanuddin University, Makassar, South Sulawesi, Indonesia; 2 Aquatic Resources Management Study Program, Hasanuddin University, Makassar, South Sulawesi, Indonesia. Corresponding author: S. B. A. Omar, [email protected] Abstract. Painted rock lobster (Panulirus versicolor) is a superior commodity with an important economic value, also being the most dominant species found in the waters of the Sorong region. Sampling was conducted from March to August 2020 in Tanjung Kasuari waters, Sorong City and Makbon waters, Sorong Regency. During the study, 365 lobster samples were captured from Tanjung Kasuari waters and 460 from Makbon waters. The results showed that in the waters of Tanjung Kasuari male lobsters had a hypoallometric growth pattern and female lobsters had an isometric growth pattern. The mean condition factors were 1.0049 for males and 0.9865 for females. On the other hand, both male and female lobsters in Makbon waters have a hypoallometric growth pattern with a mean condition factor of 0.9948 for males and 0.9875 for females. Key Words: spiny lobster, growth pattern, hypoallometric. Introduction. Lobster is a leading export commodity that has important economic value and has significantly contributed to the country's fishery sector and foreign exchange (Hasrun & Kasmawati 2018). Lobster is included in the genus Panulirus which has 21 species, 7 of which are found in Indonesian waters (Holthuis 1991; Chan 1998, 2010; Chang et al 2010; Ernawaty et al 2014; Wardiatno et al 2016; Wahyudin et al 2017a,b): P. -

Species Acromycter Nezumi (Asano, 1958)
FAMILY Congridae Kaup, 1856 - conger eels SUBFAMILY Congrinae Kaup, 1856 - conger eels [=Atteridi, Ophioides, Leptocephalini, Congeridae, Macrocephenchelyidae, Scalanogoinae, Urocongrinae] GENUS Acromycter Smith & Kanazawa, 1977 - conger eels Species Acromycter alcocki (Gilbert & Cramer, 1897) - Alcock's congrid eel Species Acromycter atlanticus Smith, 1989 - Atlantic conger eel Species Acromycter longipectoralis Karmovskaya, 2004 - longfinned conger eel Species Acromycter nezumi (Asano, 1958) - Nezumi's conger eel Species Acromycter perturbator (Parr, 1932) - perturbator conger eel GENUS Bassanago Whitley, 1948 - conger eels Species Bassanago albescens (Barnard, 1923) - hairy conger Species Bassanago bulbiceps Whitley, 1948 - swollen-headed conger eel Species Bassanago hirsutus (Castle, 1960) - deepsea conger Species Bassanago nielseni (Karmovskaya, 1990) - Nielsen's conger eel GENUS Bathycongrus Ogilby, 1898 - conger eels [=Congrina, Microcephalocongrus, Pseudoxenomystax, Rhechias, Uranoconger] Species Bathycongrus aequoreus (Gilbert & Cramer, 1897) - Kaiwi bathycongrus [=armiger] Species Bathycongrus bertini (Poll, 1953) - Poll's conger Species Bathycongrus bleekeri Fowler, 1934 - Bleeker's bathycongrus Species Bathycongrus bullisi (Smith & Kanazawa, 1977) - bullish conger Species Bathycongrus dubius (Breder, 1927) - dubius conger Species Bathycongrus guttulatus (Günther, 1887) - lined conger [=roosendaali, stimpsoni] Species Bathycongrus longicavis Karmovskaya, 2009 - Vanuatu conger Species Bathycongrus macrocercus (Alcock, 1894) - -

Informe-Taller-Peces.Pdf
Taller Regional de Evaluación del Estado de Conservación de Especies para el Mar Patagónico según criterios de la Lista Roja de UICN: PECES ÓSEOS. Buenos Aires, ARGENTINA Diciembre 2019 Results of the 2019 IUCN Regional Red List Workshop for Species of the Patagonian Sea: BONY FISHES. Agosto 2020 Con el apoyo de: 1 EXPERTOS: Claudio Buratti INIDEP- Argentina Juan Martín Díaz de Astarloa IIMyC UNMdP CONICET Mathías Hüne Fundación Ictiológica Chile Alejo Irigoyen CENPAT CONICET Mauricio Landaeta Universidad de Valparaíso Chile Cecilia Riestra INIDEP Argentina Joâo Vieira Fundacao Universidade do Río Grande do Sul - Brasil COLABORADORES EXPERTOS: Juan Martín Díaz de Astarloa y Mathías Hüne EXPERTOS IUCN: Christi Linardich REVISION Y EDICIÓN: Christi Linardich y Valeria Falabella DISEÑO Y ARTE Victoria Zavattieri Wildlife Conservation Society DIRECCIÓN: Claudio Campagna Wildlife Conservation Society COORDINACIÓN: Valeria Falabella Wildlife Conservation Society CITA: Buratti, C., Díaz de Astarloa, J.M., Falabella, V., Hüne, M., Irigoyen, Al; Landaeta, M., Linardich, C., Riestra, C., Vieira, J. Campagna, C. 2020. Informe del Taller Regional de Evaluación del Estado de Conservación de Especies para el Mar Patagónico según criterios de la Lista Roja de UICN: Peces óseos. Foro para la Conservación del Mar Patagónico y áreas de influencia. 117 pp. Citation: Buratti, C., Díaz de Astarloa, J.M., Falabella, V., Hüne, M., Irigoyen, Al; Landaeta, M., Linardich, C., Riestra, C., Vieira, J. Campagna, C. 2020. Informe del Taller Regional de Evaluación del Estado de Conservación de Especies para el Mar Patagónico según criterios de la Lista Roja de UICN: Peces óseos. Foro para la Conservación del Mar Patagónico y áreas de influencia. -

A Biophysical Profile of the Tristan Da Cunha Archipelago (PDF)
A biophysical profile of Tristan da Cunha -Sue Scott - 2017 A Biophysical Profile of the Tristan da Cunha Archipelago Sue Scott Commissioned and reviewed by The Pew Charitable Trusts 2017 1 A biophysical profile of Tristan da Cunha -Sue Scott - 2017 Contents 1. Key information on Tristan da Cunha 7 2. Geophysical setting 11 2.1. Isolated islands 11 2.2. Geology and topography 11 2.3. Climate 17 3. The ecology and biodiversity of the marine ecosystems of Tristan da Cunha 21 3.1. The pelagic ecosystem 21 3.1.1. Oceanography and ecosystem productivity 21 3.1.2. Plankton and pelagic life 25 3.1.3. Pelagic fishes 27 3.2. Marine biological surveys 29 3.3. The coastal environment 32 3.3.1. The seabed 32 3.3.2. Tides 34 3.3.3. Seashore 35 3.3.4. Intertidal bedrock and stable boulders 35 3.3.5. Rock pools and channels 39 3.3.6. Boulder beaches 42 3.4. Shallow subtidal to 40m depth 43 3.5. Deeper subtidal 45 3.6. Deep sea 48 3.7. Diversity and biogeography of benthic biota 53 3.7.1. Diversity and biogeography of fish communities 53 3.7.2. Diversity and biogeography of invertebrates 57 3.7.3. Diversity and biogeography of macroalgae 60 4. Marine mammals of Tristan da Cunha 67 4.1. Introduction 67 4.2. Seals (order Carnivora) 68 4.2.1. Eared Seals (family Otariidae) 68 4.2.2 True Seals (family Phocidae) 70 4.2.3. Rare vagrant seals 72 4.3.