Experimental Chemistry I
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
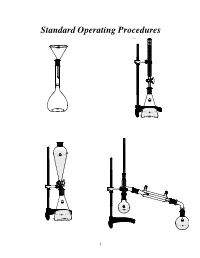
Standard Operating Procedures
Standard Operating Procedures 1 Standard Operating Procedures OVERVIEW In the following laboratory exercises you will be introduced to some of the glassware and tech- niques used by chemists to isolate components from natural or synthetic mixtures and to purify the individual compounds and characterize them by determining some of their physical proper- ties. While working collaboratively with your group members you will become acquainted with: a) Volumetric glassware b) Liquid-liquid extraction apparatus c) Distillation apparatus OBJECTIVES After finishing these sessions and reporting your results to your mentor, you should be able to: • Prepare solutions of exact concentrations • Separate liquid-liquid mixtures • Purify compounds by recrystallization • Separate mixtures by simple and fractional distillation 2 EXPERIMENT 1 Glassware Calibration, Primary and Secondary Standards, and Manual Titrations PART 1. Volumetric Glassware Calibration Volumetric glassware is used to either contain or deliver liquids at a specified temperature. Glassware manufacturers indicate this by inscribing on the volumetric ware the initials TC (to contain) or TD (to deliver) along with the calibration temperature, which is usually 20°C1. Volumetric glassware must be scrupulously clean before use. The presence of streaks or droplets is an indication of the presence of a grease film. To eliminate grease from glassware, scrub with detergent solution, rinse with tap water, and finally rinse with a small portion of distilled water. Volumetric flasks (TC) A volumetric flask has a large round bottom with only one graduation mark positioned on the long narrow neck. Graduation Mark Stopper The position of the mark facilitates the accurate and precise reading of the meniscus. If the flask is used to prepare a solution starting with a solid compound, add small amounts of sol- vent until the entire solid dissolves. -

A Simple Spectrophotometric Method for the Determination Of
J Clin Pathol: first published as 10.1136/jcp.14.2.202 on 1 March 1961. Downloaded from 202 Technical methods usual laboratory filters may give falsely increased values.) Of the clear and colourless filtrate 10 ml. is pipetted into A a 25 ml. measuring flask and 10 ml. reagent (3) added. simple spectrophotometric After mixing dilute with distilled water to the mark and method for the determination of renew mixing. After 10 minutes the mixture is ready for reading. The colour is stable, according to Watt and urea in blood and urine Chrisp, for 11 days. READING T. K. WITH, TOVE DREYER PETERSEN, AND BIRGIT PETERSEN From the Central Laboratory, Svend- The reactions are read in a spectrophotometer at 420 nm. borg County Hospital, Denmark with a blank prepared in the same way as the reaction mixture but with 3 ml. of distilled water instead of 3 ml. of blood. The urea standard (reagent 4) is treated in the Several methods for urea in determining blood and urine same way as the blood. Cuvettes of 1 cm. thickness are are in use in clinical laboratories, but none is ideal in suitable. mass routine The analysis. spectrophotometric method If p, s, and b are the extinctions of the the based on a modified Ehrlich sample, reagent proposed by Watt standard, and the blank respectively, the urea concen- and Chrisp (1954) for pure solutions is therefore of tration is interest because it can be modified for use in mass clinical analyses of blood and urine. Brown (1959) has l00(p-b)/(s-b) mg./100 ml. -

SC-300 Pro Kit Manual 1 Version 2.3D Materials Provided in the Kit
Vinmetrica SC-300 Pro Kit™ User Manual Vinmetrica SC-300 Pro Kit is a simple and robust device that provides high accuracy in determination of sulfite (SO2), pH and titratable acidity (TA) levels in wines, ciders, and other liquids. These are essential parameters to control in the effort to make high quality wines. The Pro kit includes lab accessories for the SC-300 Analyzer. Table of Contents: Materials Provided in the Kit............................................................................................................... 2 Things you will need............................................................................................................................ 2 Why Test for SO2, pH and TA?............................................................................................................ 3 Theory of Operation............................................................................................................................. 4 Setup.................................................................................................................................................... 5 Setting up the SC-300 for the first time................................................................................... 5 Assembling the Pro Kit Equipment......................................................................................... 6 Burette Maintenance................................................................................................................ 8 Burette Reading ...................................................................................................................... -

Using Serial Dilution to Understand Ppm/Ppb
Using Serial Dilution to Understand ppm/ppb Adapted from Investigating Groundwater: The Fruitvale Story, Science Education for Public Understanding Program (SEPUP), Lawrence Hall of Science (1996) by Dana Haine, UNC Superfund Research Program. Overview: Students will perform a serial dilution of food coloring to create highly diluted solutions to learn about the units, parts per million (ppm) and parts per billion (ppb), that scientists often use to describe chemical contamination of water or soil. Objectives: At the end of this activity, students will be able to: • Define serial dilution; • Define ppm and ppb; • Observe that a contaminant can be present in water even if it isn’t visible. Alignment to North Carolina Essential Standards for Science This lesson addresses components of the specific learning objectives: 8th Grade Science 8.E.1.3 Predict the safety and potability of water supplies in North Carolina based on physical and biological factors, including temperature, dissolved oxygen, pH, nitrates and phosphates, turbidity, bio-indicators 8.E.1.4 Conclude that the good health of humans requires: monitoring of the hydrosphere, water quality standards, methods of water treatment, maintaining safe water quality, stewardship Earth and Environmental Science EEn.2.4.1 Evaluate human influences on freshwater availability. EEn.2.4.2 Evaluate human influences on water quality in North Carolina’s river basins, wetlands and tidal environments. Physical Science PSc.2.1.2 Explain the phases of matter and the physical changes that matter undergoes. Materials: Student Data Collection Sheet, one per student Plastic tray with at least 9 wells or 9 test tubes, one tray/set of tubes per group White scrap paper to place under trays/tubes to observe results, one per group Red Food Coloring Two small cups of water, one labeled “rinse” Medicine dropper, one per group Paper towels Red colored pencils (optional) Duration 20-30 minutes Procedure: 1. -

A Fiber Optic Ammonia Sensor Using a Universal Ph Indicator
Sensors 2014, 14, 4060-4073; doi:10.3390/s140304060 OPEN ACCESS sensors ISSN 1424-8220 www.mdpi.com/journal/sensors Article A Fiber Optic Ammonia Sensor Using a Universal pH Indicator Adolfo J. Rodríguez 1,*, Carlos R. Zamarreño 2, Ignacio R. Matías 2, Francisco. J. Arregui 2, Rene F. Domínguez Cruz 1 and Daniel. A. May-Arrioja 1 1 Fiber and Integrated Optics Laboratory, Electronics Engineering Department, UAM Reynosa Rodhe, Universidad Autónoma de Tamaulipas, Carr. Reynosa-San Fernando S/N, Reynosa, Tamaulipas 88779, Mexico; E-Mails: [email protected] (R.F.D.C.); [email protected] (D.A.M.-A.) 2 Departamento de Ingeniería Eléctrica y Electrónica, Universidad Pública de Navarra, Edif. Los Tejos, Campus Arrosadia, Pamplona España 31006, Spain; E-Mails: [email protected] (C.R.Z.); [email protected] (I.R.M.); [email protected] (F.J.A.) * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel.: +52-899-9213300 (ext. 8315); Fax: +52-899-921-3300 (ext. 8050). Received: 27 December 2013; in revised form: 12 February 2014 / Accepted: 18 February 2014 / Published: 27 February 2014 Abstract: A universal pH indicator is used to fabricate a fiber optic ammonia sensor. The advantage of this pH indicator is that it exhibits sensitivity to ammonia over a broad wavelength range. This provides a differential response, with a valley around 500 nm and a peak around 650 nm, which allows us to perform ratiometric measurements. The ratiometric measurements provide not only an enhanced signal, but can also eliminate any external disturbance due to humidity or temperature fluctuations. -

General Chemistry Laboratory I Manual
GENERAL CHEMISTRY LABORATORY I MANUAL Fall Semester Contents Laboratory Equipments .............................................................................................................................. i Experiment 1 Measurements and Density .............................................................................................. 10 Experiment 2 The Stoichiometry of a Reaction ..................................................................................... 31 Experiment 3 Titration of Acids and Bases ............................................................................................ 10 Experiment 4 Oxidation – Reduction Titration ..................................................................................... 49 Experiment 5 Quantitative Analysis Based on Gas Properties ............................................................ 57 Experiment 6 Thermochemistry: The Heat of Reaction ....................................................................... 67 Experiment 7 Group I: The Soluble Group ........................................................................................... 79 Experiment 8 Gravimetric Analysis ........................................................................................................ 84 Scores of the General Chemistry Laboratory I Experiments ............................................................... 93 LABORATORY EQUIPMENTS BEAKER (BEHER) Beakers are containers which can be used for carrying out reactions, heating solutions, and for water baths. They are for -

Laboratory Exercises in Microbiology: Discovering the Unseen World Through Hands-On Investigation
City University of New York (CUNY) CUNY Academic Works Open Educational Resources Queensborough Community College 2016 Laboratory Exercises in Microbiology: Discovering the Unseen World Through Hands-On Investigation Joan Petersen CUNY Queensborough Community College Susan McLaughlin CUNY Queensborough Community College How does access to this work benefit ou?y Let us know! More information about this work at: https://academicworks.cuny.edu/qb_oers/16 Discover additional works at: https://academicworks.cuny.edu This work is made publicly available by the City University of New York (CUNY). Contact: [email protected] Laboratory Exercises in Microbiology: Discovering the Unseen World through Hands-On Investigation By Dr. Susan McLaughlin & Dr. Joan Petersen Queensborough Community College Laboratory Exercises in Microbiology: Discovering the Unseen World through Hands-On Investigation Table of Contents Preface………………………………………………………………………………………i Acknowledgments…………………………………………………………………………..ii Microbiology Lab Safety Instructions…………………………………………………...... iii Lab 1. Introduction to Microscopy and Diversity of Cell Types……………………......... 1 Lab 2. Introduction to Aseptic Techniques and Growth Media………………………...... 19 Lab 3. Preparation of Bacterial Smears and Introduction to Staining…………………...... 37 Lab 4. Acid fast and Endospore Staining……………………………………………......... 49 Lab 5. Metabolic Activities of Bacteria…………………………………………….…....... 59 Lab 6. Dichotomous Keys……………………………………………………………......... 77 Lab 7. The Effect of Physical Factors on Microbial Growth……………………………... 85 Lab 8. Chemical Control of Microbial Growth—Disinfectants and Antibiotics…………. 99 Lab 9. The Microbiology of Milk and Food………………………………………………. 111 Lab 10. The Eukaryotes………………………………………………………………........ 123 Lab 11. Clinical Microbiology I; Anaerobic pathogens; Vectors of Infectious Disease….. 141 Lab 12. Clinical Microbiology II—Immunology and the Biolog System………………… 153 Lab 13. Putting it all Together: Case Studies in Microbiology…………………………… 163 Appendix I. -

Laboratory Equipment Reference Sheet
Laboratory Equipment Stirring Rod: Reference Sheet: Iron Ring: Description: Glass rod. Uses: To stir combinations; To use in pouring liquids. Evaporating Dish: Description: Iron ring with a screw fastener; Several Sizes Uses: To fasten to the ring stand as a support for an apparatus Description: Porcelain dish. Buret Clamp/Test Tube Clamp: Uses: As a container for small amounts of liquids being evaporated. Glass Plate: Description: Metal clamp with a screw fastener, swivel and lock nut, adjusting screw, and a curved clamp. Uses: To hold an apparatus; May be fastened to a ring stand. Mortar and Pestle: Description: Thick glass. Uses: Many uses; Should not be heated Description: Heavy porcelain dish with a grinder. Watch Glass: Uses: To grind chemicals to a powder. Spatula: Description: Curved glass. Uses: May be used as a beaker cover; May be used in evaporating very small amounts of Description: Made of metal or porcelain. liquid. Uses: To transfer solid chemicals in weighing. Funnel: Triangular File: Description: Metal file with three cutting edges. Uses: To scratch glass or file. Rubber Connector: Description: Glass or plastic. Uses: To hold filter paper; May be used in pouring Description: Short length of tubing. Medicine Dropper: Uses: To connect parts of an apparatus. Pinch Clamp: Description: Glass tip with a rubber bulb. Uses: To transfer small amounts of liquid. Forceps: Description: Metal clamp with finger grips. Uses: To clamp a rubber connector. Test Tube Rack: Description: Metal Uses: To pick up or hold small objects. Beaker: Description: Rack; May be wood, metal, or plastic. Uses: To hold test tubes in an upright position. -

Determination of the Solubility Product of an Ionic Compound
Determination of the Solubility Product of an Ionic Compound Kyle Miller January 11, 2007 1 Data The following data were collected. Dilution Precipitation well Ca2+ #6 OH− #4 2 Calculations 2.1 Calcium Ion Serial Dilution The first well without a precipitate is well number 6. The concentration of calcium ions per well is 0.10 [Ca2+] = M (1) 2n where n is the well number. So, the concentration of calcium ions in well number 6 is 0.10 −3 26 M = 1.6 × 10 M For each of the wells in this series, the concentration of added NaOH was a constant 0.1 M 2 = 0.05 M after being added to each well. 2+ − The Ksp for this ion equilibrium, Ca(OH)2 Ca + 2OH , is 2+ −2 Ksp = Ca OH (2) So, putting in the concentrations for well number 6, which is assumed to have complete −3 2 −6 dissociation of the calcium hydroxide, Ksp = 1.6 × 10 [0.05] = 3.9 × 10 1 2.2 Hydroxide Ion Serial Dilution The first well without precipitate for this series is well number 4. The concentration of hydroxide ions per well is 0.10 [OH−] = M (3) 2n again, with n being the well number. So, the concentration of hydroxide ions in well 0.10 −3 number 4 is 24 M = 6.3 × 10 M For each of the wells in this series, the concentration of added calcium nitrate was a constant 0.1 M 2 = 0.05 M after being added to each well. Using the ion equilibrium equation and these concentrations, we can find that Ksp = [0.05] 6.3 × 10−32 = 2.0 × 10−6 ¯ 3.9×10−6+2.0×10−6 −6 Then, Ksp = 2 = 3.0 × 10 −6 The actual Ksp is 5.02×10 and the percent error from this value to the calculated average |3.0×10−6−5.02×10−6| is 5.02×10−6 = 40.% 3 Discussion 1. -

Experiment 16
FV 2/10/2017 Experiment 16 HYDRONIUM ION CONCENTRATION MATERIALS: 7 centrifuge tubes with 10 mL mark, six 25 x 150 mm large test tubes, 5 mL pipet, 50 mL beaker, 50 mL graduated cylinder, 1.0 M HCl, 1.0 M CH3COOH, CH3COONa, methyl violet and methyl orange indicator solutions, test tube rack, calibrated pH meter (instructor use only). PURPOSE: The purpose of this experiment is to perform serial dilutions and use indicators to estimate the pH of various solutions. LEARNING OBJECTIVES: By the end of this experiment, the students should be able to demonstrate the following proficiencies: 1. Prepare solutions by serial dilution. + 2. Correlate the H3O ion concentration of a solution with its pH value. 3. Use indicators to estimate the pH of solutions of various acid concentrations. 4. Explain the common ion effect. 5. Calculate percent dissociation of a weak acid. PRE-LAB: Read over the experiment and complete the pre-lab questions on OWL before the lab. DISCUSSION: In any aqueous solution, the product of the hydronium ion concentration and the hydroxide ion concentration is equal to a constant, known as Kw. At a temperature of 25°C, this product is: + - -14 Kw = [H3O ]⋅ [OH ] = 1.0 x 10 (25° C) + - If the [H3O ] concentration is altered, the [OH ] concentration changes so that the product of the two terms remains 1.0 x 10-14. In pure, distilled water at 25°C: + - -7 [H3O ] = [OH ] = 1.0 x 10 M (25° C) To describe acidity in a simple way, the pH scale was adopted: + pH = -log[H3O ] Thus, for pure, distilled water at 25°C: -7 = ° pH = -log(1.0 x 10 ) 7.00 (25 C) + The practical range of pH in aqueous solutions at 25°C is from 0 to 14. -

Kjeldahl Distillation Unit
Terms of Business v There are frequent fluctuations in the cost of input. So we may not be able to give prior intimation regarding changes in the price structure. v Packing, Forwarding, Freight and Insurance will be Charged Extra. v Sales Tax and Excise Duty will be extra as per Central or State Government Rules. v Every possible care is taken in packing, but it is difficult to undertake responsibility of loss, breakage or damage during transit. v R/R and our bill be sent though V. P. P. or any scheduled bank. New customers are requested to advance 25% of the estimated value. v While placing the order, please give clear despatch instructions indicating C. S. T. Number, Name of Railway Station, Your Banker and Our Catalogue Number. v All problems shall be solved amicably subject to Ambala Jurisdiction. GUPTA SCIENTIFIC INDUSTRIES Ambala Cantt. THIS CANCELS OUR ALL PREVIOUS PRICE LISTS PPPrice List 2017-18 CATALOGUE CONTENTS Product Range Pages Interchangeable Standard Joints, Adapters, Stirrers, Condensers, 1 to 49 Laboratory Flasks, Columns, Separating Funnels, Assemblies, Glasswares Water Distillation, Essential Oil, Soxhlet Apparatus. Volumetric Burettes, Pipettes, Micro Pipettes, Measuring Cylinders, 50 to 74 Glasswares Measuring Flasks, Culture Tubes, Centrifuge Tube & Test Tubes. Sintered Sintered Crucibles, Funnels, Chromatography Columns and 75 to 84 Glasswares Filter Assembly. Gas Estimation Impinger, Gas Burettes, Tubes, Gas Wash Bottles, 85 to 90 Apparatus Oxygen Purity and Orsat Apparatus. Special Semi Micro Ware, Clinical Laboratory Apparatus, 91 to 97 Glasswares Electrodes and Milk Testing Apparatus. Supplementary Stopcocks, Rotaflow Screw Type Stopcocks, Teflon Key 98 to 123 Glasswares Stopcocks, Manometers, Weighing Bottles, S. -

A Study of Form and Content for a Laboratory Manual to Be Used by Students in General Chemistry Laboratory
Brigham Young University BYU ScholarsArchive Theses and Dissertations 1946-01-01 A study of form and content for a laboratory manual to be used by students in general chemistry laboratory Berne P. Broadbent Brigham Young University - Provo Follow this and additional works at: https://scholarsarchive.byu.edu/etd BYU ScholarsArchive Citation Broadbent, Berne P., "A study of form and content for a laboratory manual to be used by students in general chemistry laboratory" (1946). Theses and Dissertations. 8176. https://scholarsarchive.byu.edu/etd/8176 This Thesis is brought to you for free and open access by BYU ScholarsArchive. It has been accepted for inclusion in Theses and Dissertations by an authorized administrator of BYU ScholarsArchive. For more information, please contact [email protected], [email protected]. ?_(j;, . , i. ~ ~ (12 ' -8?5: 11% -· • A STUDY OF FOFM CONTENTFOR A LABORATORY -\ .AND MANUALTO BE USED BY STUDENTSIN GENERALCEEi/IISTRY LABORATORY' A THESIS SUBMITTEDTO \ THE DEPAR'.I3\t1Ell."'T OF CHEMISTRY OF ··! BRIGHAMYOUNG UNIVERSITY,. IN PARTIALFULFII.lllENT OF THEREQ,UIREMENTS·FOR THE DEGREE OF MASTEROF SCIENCE ... .,; . •·' .. ...• • .• . • "f ... ·.. .. ,. ·: :. !./:.•:-.:.lo>•.-,:... ... ... ..........• • • • p ,.. .,• • • ...• • . ~. ••,,. ................. :... ~•••,,.c • ..............• • • • • • .. f" ·~•-~-·"••• • • • ... • .., : :·.•··•:'"'•••:'"',. ·.-··.::· 147141 BY BERNEP. BROADBENT . " 1946 .,_ - ii \ ., This Thesis by Berne P.- Broadbent is accepted in 1ts P:esent form by the Departm·ent of Chem�stry as satisfying the Thesis requirement.for the degree of .J Master of Science • ,• . - .} .. iii PREF.ACE The constantly broadening field assigned to general chemistry demands that material be carefully selected and that ever increasing attention be given to preparing this material and presenting it_ to the student.- The following study was made to develop a laboratory manual that would increase the effectiveness of laboratory work.