THEORY of PROTEIN FOLDING: the Energy Landscape Perspective
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

POLYMER STRUCTURE and CHARACTERIZATION Professor
POLYMER STRUCTURE AND CHARACTERIZATION Professor John A. Nairn Fall 2007 TABLE OF CONTENTS 1 INTRODUCTION 1 1.1 Definitions of Terms . 2 1.2 Course Goals . 5 2 POLYMER MOLECULAR WEIGHT 7 2.1 Introduction . 7 2.2 Number Average Molecular Weight . 9 2.3 Weight Average Molecular Weight . 10 2.4 Other Average Molecular Weights . 10 2.5 A Distribution of Molecular Weights . 11 2.6 Most Probable Molecular Weight Distribution . 12 3 MOLECULAR CONFORMATIONS 21 3.1 Introduction . 21 3.2 Nomenclature . 23 3.3 Property Calculation . 25 3.4 Freely-Jointed Chain . 27 3.4.1 Freely-Jointed Chain Analysis . 28 3.4.2 Comment on Freely-Jointed Chain . 34 3.5 Equivalent Freely Jointed Chain . 37 3.6 Vector Analysis of Polymer Conformations . 38 3.7 Freely-Rotating Chain . 41 3.8 Hindered Rotating Chain . 43 3.9 More Realistic Analysis . 45 3.10 Theta (Θ) Temperature . 47 3.11 Rotational Isomeric State Model . 48 4 RUBBER ELASTICITY 57 4.1 Introduction . 57 4.2 Historical Observations . 57 4.3 Thermodynamics . 60 4.4 Mechanical Properties . 62 4.5 Making Elastomers . 68 4.5.1 Diene Elastomers . 68 0 4.5.2 Nondiene Elastomers . 69 4.5.3 Thermoplastic Elastomers . 70 5 AMORPHOUS POLYMERS 73 5.1 Introduction . 73 5.2 The Glass Transition . 73 5.3 Free Volume Theory . 73 5.4 Physical Aging . 73 6 SEMICRYSTALLINE POLYMERS 75 6.1 Introduction . 75 6.2 Degree of Crystallization . 75 6.3 Structures . 75 Chapter 1 INTRODUCTION The topic of polymer structure and characterization covers molecular structure of polymer molecules, the arrangement of polymer molecules within a bulk polymer material, and techniques used to give information about structure or properties of polymers. -

Ideal Chain Conformations and Statistics
ENAS 606 : Polymer Physics Chinedum Osuji 01.24.2013:HO2 Ideal Chain Conformations and Statistics 1 Overview Consideration of the structure of macromolecules starts with a look at the details of chain level chemical details which can impact the conformations adopted by the polymer. In the case of saturated carbon ◦ backbones, while maintaining the desired Ci−1 − Ci − Ci+1 bond angle of 112 , the placement of the final carbon in the triad above can occur at any point along the circumference of a circle, defining a torsion angle '. We can readily recognize the energetic differences as a function this angle, U(') such that there are 3 minima - a deep minimum corresponding the the trans state, for which ' = 0 and energetically equivalent gauche− and gauche+ states at ' = ±120 degrees, as shown in Fig 1. Figure 1: Trans, gauche- and gauche+ configurations, and their energetic sates 1.1 Static Flexibility The static flexibility of the chain in equilibrium is determined by the difference between the levels of the energy minima corresponding the gauche and trans states, ∆. If ∆ < kT , the g+, g− and t states occur with similar probability, and so the chain can change direction and appears as a random coil. If ∆ takes on a larger value, then the t conformations will be enriched, so the chain will be rigid locally, but on larger length scales, the eventual occurrence of g+ and g− conformations imparts a random conformation. Overall, if we ignore details on some length scale smaller than lp, the persistence length, the polymer appears as a continuous flexible chain where 1 lp = l0 exp(∆/kT ) (1) where l0 is something like a monomer length. -

Native-Like Mean Structure in the Unfolded Ensemble of Small Proteins
B doi:10.1016/S0022-2836(02)00888-4 available online at http://www.idealibrary.com on w J. Mol. Biol. (2002) 323, 153–164 Native-like Mean Structure in the Unfolded Ensemble of Small Proteins Bojan Zagrovic1, Christopher D. Snow1, Siraj Khaliq2 Michael R. Shirts2 and Vijay S. Pande1,2* 1Biophysics Program The nature of the unfolded state plays a great role in our understanding of Stanford University, Stanford proteins. However, accurately studying the unfolded state with computer CA 94305-5080, USA simulation is difficult, due to its complexity and the great deal of sampling required. Using a supercluster of over 10,000 processors we 2Department of Chemistry have performed close to 800 ms of molecular dynamics simulation in Stanford University, Stanford atomistic detail of the folded and unfolded states of three polypeptides CA 94305-5080, USA from a range of structural classes: the all-alpha villin headpiece molecule, the beta hairpin tryptophan zipper, and a designed alpha-beta zinc finger mimic. A comparison between the folded and the unfolded ensembles reveals that, even though virtually none of the individual members of the unfolded ensemble exhibits native-like features, the mean unfolded structure (averaged over the entire unfolded ensemble) has a native-like geometry. This suggests several novel implications for protein folding and structure prediction as well as new interpretations for experiments which find structure in ensemble-averaged measurements. q 2002 Elsevier Science Ltd. All rights reserved Keywords: mean-structure hypothesis; unfolded state of proteins; *Corresponding author distributed computing; conformational averaging Introduction under folding conditions, with some notable exceptions.14 – 16 This is understandable since under Historically, the unfolded state of proteins has such conditions the unfolded state is an unstable, received significantly less attention than the folded fleeting species making any kind of quantitative state.1 The reasons for this are primarily its struc- experimental measurement very difficult. -

DNA Energy Landscapes Via Calorimetric Detection of Microstate Ensembles of Metastable Macrostates and Triplet Repeat Diseases
DNA energy landscapes via calorimetric detection of microstate ensembles of metastable macrostates and triplet repeat diseases Jens Vo¨ lkera, Horst H. Klumpb, and Kenneth J. Breslauera,c,1 aDepartment of Chemistry and Chemical Biology, Rutgers, The State University of New Jersey, 610 Taylor Rd, Piscataway, NJ 08854; bDepartment of Molecular and Cell Biology, University of Cape Town, Private Bag, Rondebosch 7800, South Africa; and cCancer Institute of New Jersey, New Brunswick, NJ 08901 Communicated by I. M. Gelfand, Rutgers, The State University of New Jersey, Piscataway, NJ, October 15, 2008 (received for review September 8, 2008) Biopolymers exhibit rough energy landscapes, thereby allowing biological role(s), if any, of kinetically stable (metastable) mi- biological processes to access a broad range of kinetic and ther- crostates that make up the time-averaged, native state ensembles modynamic states. In contrast to proteins, the energy landscapes of macroscopic nucleic acid states remains to be determined. of nucleic acids have been the subject of relatively few experimen- As part of an effort to address this deficiency, we report here tal investigations. In this study, we use calorimetric and spectro- experimental evidence for the presence of discrete microstates scopic observables to detect, resolve, and selectively enrich ener- in metastable triplet repeat bulge looped ⍀-DNAs of potential getically discrete ensembles of microstates within metastable DNA biological significance. The specific triplet repeat bulge looped structures. Our results are consistent with metastable, ‘‘native’’ ⍀-DNA species investigated mimic slipped DNA structures DNA states being composed of an ensemble of discrete and corresponding to intermediates in the processes that lead to kinetically stable microstates of differential stabilities, rather than DNA expansion in triplet repeat diseases (36). -
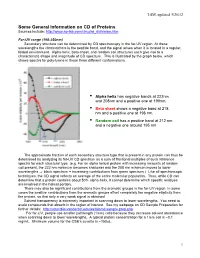
CD of Proteins Sources Include
LSM, updated 3/26/12 Some General Information on CD of Proteins Sources include: http://www.ap-lab.com/circular_dichroism.htm Far-UV range (190-250nm) Secondary structure can be determined by CD spectroscopy in the far-UV region. At these wavelengths the chromophore is the peptide bond, and the signal arises when it is located in a regular, folded environment. Alpha-helix, beta-sheet, and random coil structures each give rise to a characteristic shape and magnitude of CD spectrum. This is illustrated by the graph below, which shows spectra for poly-lysine in these three different conformations. • Alpha helix has negative bands at 222nm and 208nm and a positive one at 190nm. • Beta sheet shows a negative band at 218 nm and a positive one at 196 nm. • Random coil has a positive band at 212 nm and a negative one around 195 nm. The approximate fraction of each secondary structure type that is present in any protein can thus be determined by analyzing its far-UV CD spectrum as a sum of fractional multiples of such reference spectra for each structural type. (e.g. For an alpha helical protein with increasing amounts of random coil present, the 222 nm minimum becomes shallower and the 208 nm minimum moves to lower wavelengths ⇒ black spectrum + increasing contributions from green spectrum.) Like all spectroscopic techniques, the CD signal reflects an average of the entire molecular population. Thus, while CD can determine that a protein contains about 50% alpha-helix, it cannot determine which specific residues are involved in the helical portion. -

Ruggedness in the Free Energy Landscape Dictates Misfolding of the Prion Protein
Article Ruggedness in the Free Energy Landscape Dictates Misfolding of the Prion Protein Roumita Moulick 1, Rama Reddy Goluguri 1 and Jayant B. Udgaonkar 1,2 1 - National Centre for Biological Sciences, Tata Institute of Fundamental Research, Bengaluru 560065, India 2 - Indian Institute of Science Education and Research, Pashan, Pune 411008, India Correspondence to Jayant B. Udgaonkar: Indian Institute of Science Education and Research, Pashan, Pune 411008, India. [email protected] https://doi.org/10.1016/j.jmb.2018.12.009 Edited by Sheena Radford Abstract Experimental determination of the key features of the free energy landscapes of proteins, which dictate their adeptness to fold correctly, or propensity to misfold and aggregate and which are modulated upon a change from physiological to aggregation-prone conditions, is a difficult challenge. In this study, sub-millisecond kinetic measurements of the folding and unfolding of the mouse prion protein reveal how the free energy landscape becomes more complex upon a shift from physiological (pH 7) to aggregation-prone (pH 4) conditions. Folding and unfolding utilize the same single pathway at pH 7, but at pH 4, folding occurs on a pathway distinct from the unfolding pathway. Moreover, the kinetics of both folding and unfolding at pH 4 depend not only on the final conditions but also on the conditions under which the processes are initiated. Unfolding can be made to switch to occur on the folding pathway by varying the initial conditions. Folding and unfolding pathways appear to occupy different regions of the free energy landscape, which are separated by large free energy barriers that change with a change in the initial conditions. -

Large-Scale Analyses of Site-Specific Evolutionary Rates Across
G C A T T A C G G C A T genes Article Large-Scale Analyses of Site-Specific Evolutionary Rates across Eukaryote Proteomes Reveal Confounding Interactions between Intrinsic Disorder, Secondary Structure, and Functional Domains Joseph B. Ahrens, Jordon Rahaman and Jessica Siltberg-Liberles * Department of Biological Sciences, Florida International University, Miami, FL 33199, USA; [email protected] (J.B.A.); jraha001@fiu.edu (J.R.) * Correspondence: jliberle@fiu.edu; Tel.: +1-305-348-7508 Received: 1 October 2018; Accepted: 9 November 2018; Published: 14 November 2018 Abstract: Various structural and functional constraints govern the evolution of protein sequences. As a result, the relative rates of amino acid replacement among sites within a protein can vary significantly. Previous large-scale work on Metazoan (Animal) protein sequence alignments indicated that amino acid replacement rates are partially driven by a complex interaction among three factors: intrinsic disorder propensity; secondary structure; and functional domain involvement. Here, we use sequence-based predictors to evaluate the effects of these factors on site-specific sequence evolutionary rates within four eukaryotic lineages: Metazoans; Plants; Saccharomycete Fungi; and Alveolate Protists. Our results show broad, consistent trends across all four Eukaryote groups. In all four lineages, there is a significant increase in amino acid replacement rates when comparing: (i) disordered vs. ordered sites; (ii) random coil sites vs. sites in secondary structures; and (iii) inter-domain linker sites vs. sites in functional domains. Additionally, within Metazoans, Plants, and Saccharomycetes, there is a strong confounding interaction between intrinsic disorder and secondary structure—alignment sites exhibiting both high disorder propensity and involvement in secondary structures have very low average rates of sequence evolution. -

Introducing the Levinthal's Protein Folding Paradox and Its Solution
Article pubs.acs.org/jchemeduc Introducing the Levinthal’s Protein Folding Paradox and Its Solution Leandro Martínez* Department of Physical Chemistry, Institute of Chemistry, University of Campinas, Campinas, Saõ Paulo 13083-970, Brazil *S Supporting Information ABSTRACT: The protein folding (Levinthal’s) paradox states that it would not be possible in a physically meaningful time to a protein to reach the native (functional) conformation by a random search of the enormously large number of possible structures. This paradox has been solved: it was shown that small biases toward the native conformation result in realistic folding times of realistic-length sequences. This solution of the paradox is, however, not amenable to most chemistry or biology students due to the demanding mathematics. Here, a simplification of the study of the paradox and its solution is provided so that it is accessible to chemists and biologists at an undergraduate or graduate level. Despite its simplicity, the model captures some fundamental aspects of the protein folding mechanism and allows students to grasp the actual significance of the popular folding funnel representation of the protein energy landscape. The analysis of the folding model provides a rich basis for a discussion of the relationships between kinetics and thermodynamics on a fundamental level and on student perception of the time-scales of molecular phenomena. KEYWORDS: Upper-Division Undergraduate, Graduate Education, Biochemistry, Biophysical Chemistry, Molecular Biology, Proteins/Peptides, -

Protein Folding and Structure Prediction
Protein Folding and Structure Prediction A Statistician's View Ingo Ruczinski Department of Biostatistics, Johns Hopkins University Proteins Amino acids without peptide bonds. Amino acids with peptide bonds. ¡ Amino acids are the building blocks of proteins. Proteins Both figures show the same protein (the bacterial protein L). The right figure also highlights the secondary structure elements. Space Resolution limit of a light microscope Glucose Ribosome Red blood cell C−C bond Hemoglobin Bacterium 1 10 100 1000 10000 100000 1nm 1µm Distance [ A° ] Energy C−C bond Green Noncovalent bond light Glucose Thermal ATP 0.1 1 10 100 1000 Energy [ kcal/mol ] Non-Bonding Interactions Amino acids of a protein are joined by covalent bonding interactions. The polypep- tide is folded in three dimension by non-bonding interactions. These interactions, which can easily be disrupted by extreme pH, temperature, pressure, and denatu- rants, are: Electrostatic Interactions (5 kcal/mol) Hydrogen-bond Interactions (3-7 kcal/mol) Van Der Waals Interactions (1 kcal/mol) ¡ Hydrophobic Interactions ( 10 kcal/mol) The total inter-atomic force acting between two atoms is the sum of all the forces they exert on each other. Energy Profile Transition State Denatured State Native State Radius of Gyration of Denatured Proteins Do chemically denatured proteins behave as random coils? The radius of gyration Rg of a protein is defined as the root mean square dis- tance from each atom of the protein to their centroid. For an ideal (infinitely thin) random-coil chain in a solvent, the average radius 0.5 ¡ of gyration of a random coil is a simple function of its length n: Rg n For an excluded volume polymer (a polymer with non-zero thickness and non- trivial interactions between monomers) in a solvent, the average radius of gyra- 0.588 tion, we have Rg n (Flory 1953). -

Chapter 6 Protein Structure and Folding
Chapter 6 Protein Structure and Folding 1. Secondary Structure 2. Tertiary Structure 3. Quaternary Structure and Symmetry 4. Protein Stability 5. Protein Folding Myoglobin Introduction 1. Proteins were long thought to be colloids of random structure 2. 1934, crystal of pepsin in X-ray beam produces discrete diffraction pattern -> atoms are ordered 3. 1958 first X-ray structure solved, sperm whale myoglobin, no structural regularity observed 4. Today, approx 50’000 structures solved => remarkable degree of structural regularity observed Hierarchy of Structural Layers 1. Primary structure: amino acid sequence 2. Secondary structure: local arrangement of peptide backbone 3. Tertiary structure: three dimensional arrangement of all atoms, peptide backbone and amino acid side chains 4. Quaternary structure: spatial arrangement of subunits 1) Secondary Structure A) The planar peptide group limits polypeptide conformations The peptide group ha a rigid, planar structure as a consequence of resonance interactions that give the peptide bond ~40% double bond character The trans peptide group The peptide group assumes the trans conformation 8 kJ/mol mire stable than cis Except Pro, followed by cis in 10% Torsion angles between peptide groups describe polypeptide chain conformations The backbone is a chain of planar peptide groups The conformation of the backbone can be described by the torsion angles (dihedral angles, rotation angles) around the Cα-N (Φ) and the Cα-C bond (Ψ) Defined as 180° when extended (as shown) + = clockwise, seen from Cα Not -

Protein Misfolding in Human Diseases
Linköping Studies in Science and Technology Dissertation No. 1239 Protein Misfolding in Human Diseases Karin Almstedt Biochemistry Department of Physics, Chemistry and Biology Linköping University, SE-581 83 Linköping, Sweden Linköping 2009 The cover shows a Himalayan mountain range, symbolizing a protein folding landscape. During the course of the research underlying this thesis, Karin Almstedt was enrolled in Forum Scientium, a multidisciplinary doctoral program at Linköping University, Sweden. Copyright © 2009 Karin Almstedt ISBN: 978-91-7393-698-9 ISSN: 0345-7524 Printed in Sweden by LiU-Tryck Linköping 2009 Never Stop Exploring 83 ABSTRACT The studies in this thesis are focused on misfolded proteins involved in human disease. There are several well known diseases that are due to aberrant protein folding. These types of diseases can be divided into three main categories: 1. Loss-of-function diseases 2. Gain-of-toxic-function diseases 3. Infectious misfolding diseases Most loss-of-function diseases are caused by aberrant folding of important proteins. These proteins often misfold due to inherited mutations. The rare disease carbonic anhydrase II deficiency syndrome (CADS) can manifest in carriers of point mutations in the human carbonic anhydrase II (HCA II) gene. One mutation associated with CADS entails the His107Tyr (H107Y) substitution. We have demonstrated that the H107Y mutation is a remarkably destabilizing mutation influencing the folding behavior of HCA II. A mutational survey of position H107 and a neighboring conserved position E117 has been performed entailing the mutants H107A, H107F, H107N, E117A and the double mutants H107A/E117A and H107N/E117A. We have also shown that the binding of specific ligands can stabilize the disease causing mutant, and shift the folding equilibrium towards the native state, providing a starting point for small molecule drugs for CADS. -

Helix-Coil Conformational Change Accompanied by Anisotropic–Isotropic Transition
Polymer Journal, Vol.33, No. 11, pp 898—901 (2001) NOTES Helix-Coil Conformational Change Accompanied by Anisotropic–Isotropic Transition † Ning LIU, Jiaping LIN, Tao C HEN, Jianding CHEN, Dafei ZHOU, and Le LI Department of Polymer Science and Engineering, East China University of Science and Technology, Shanghai 200237, P. R. China (Received November 22, 2000; Accepted July 1, 2001) KEY WORDS Helix-Coil Transition / Liquid Crystal Polymer / Polypeptide / Anisotropic– Isotropic Transition / Polypeptides, particularly poly(γ-benzyl-L- limited study of this acid induced anisotropic–isotropic glutamate) (PBLG) and other glutamic acid ester transition has been reported.13, 14 The precise molecular polymers, have the ability to exist in α-helix, a well de- mechanism of the transition remains to be determined. fined chain conformation of long chain order and retain In the present work, the texture changes in the acid- such a structure in numerous organic solvents that sup- induced anisotropic–isotropic transition were studied port intramolecular hydrogen bonding.1, 2 If PBLG and by microscope observations and the corresponding related polypeptides are dissolved in a binary solvent molecular conformational changes were followed by IR mixture containing a non-helicogenic component such measurements. The effect of the polymer concentra- as dichloroacetic acid (DCA) or trifluoroacetic acid tion was studied. According to the information gained (TFA), they can undergo a conformational change from through the experiment, the molecular mechanism with the α-helix to random coil form due to the changes respect to the acid-induced helix-coil conformational in solvent composition, temperature or both.3, 4 The change was suggested.