Metagenomic/Metatranscriptomic Study of Organisms Entrapped
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

CUED Phd and Mphil Thesis Classes
High-throughput Experimental and Computational Studies of Bacterial Evolution Lars Barquist Queens' College University of Cambridge A thesis submitted for the degree of Doctor of Philosophy 23 August 2013 Arrakis teaches the attitude of the knife { chopping off what's incomplete and saying: \Now it's complete because it's ended here." Collected Sayings of Muad'dib Declaration High-throughput Experimental and Computational Studies of Bacterial Evolution The work presented in this dissertation was carried out at the Wellcome Trust Sanger Institute between October 2009 and August 2013. This dissertation is the result of my own work and includes nothing which is the outcome of work done in collaboration except where specifically indicated in the text. This dissertation does not exceed the limit of 60,000 words as specified by the Faculty of Biology Degree Committee. This dissertation has been typeset in 12pt Computer Modern font using LATEX according to the specifications set by the Board of Graduate Studies and the Faculty of Biology Degree Committee. No part of this dissertation or anything substantially similar has been or is being submitted for any other qualification at any other university. Acknowledgements I have been tremendously fortunate to spend the past four years on the Wellcome Trust Genome Campus at the Sanger Institute and the European Bioinformatics Institute. I would like to thank foremost my main collaborators on the studies described in this thesis: Paul Gardner and Gemma Langridge. Their contributions and support have been invaluable. I would also like to thank my supervisor, Alex Bateman, for giving me the freedom to pursue a wide range of projects during my time in his group and for advice. -

Pinpointing the Origin of Mitochondria Zhang Wang Hanchuan, Hubei
Pinpointing the origin of mitochondria Zhang Wang Hanchuan, Hubei, China B.S., Wuhan University, 2009 A Dissertation presented to the Graduate Faculty of the University of Virginia in Candidacy for the Degree of Doctor of Philosophy Department of Biology University of Virginia August, 2014 ii Abstract The explosive growth of genomic data presents both opportunities and challenges for the study of evolutionary biology, ecology and diversity. Genome-scale phylogenetic analysis (known as phylogenomics) has demonstrated its power in resolving the evolutionary tree of life and deciphering various fascinating questions regarding the origin and evolution of earth’s contemporary organisms. One of the most fundamental events in the earth’s history of life regards the origin of mitochondria. Overwhelming evidence supports the endosymbiotic theory that mitochondria originated once from a free-living α-proteobacterium that was engulfed by its host probably 2 billion years ago. However, its exact position in the tree of life remains highly debated. In particular, systematic errors including sparse taxonomic sampling, high evolutionary rate and sequence composition bias have long plagued the mitochondrial phylogenetics. This dissertation employs an integrated phylogenomic approach toward pinpointing the origin of mitochondria. By strategically sequencing 18 phylogenetically novel α-proteobacterial genomes, using a set of “well-behaved” phylogenetic markers with lower evolutionary rates and less composition bias, and applying more realistic phylogenetic models that better account for the systematic errors, the presented phylogenomic study for the first time placed the mitochondria unequivocally within the Rickettsiales order of α- proteobacteria, as a sister clade to the Rickettsiaceae and Anaplasmataceae families, all subtended by the Holosporaceae family. -

ID 2 | Issue No: 4.1 | Issue Date: 29.10.14 | Page: 1 of 24 © Crown Copyright 2014 Identification of Corynebacterium Species
UK Standards for Microbiology Investigations Identification of Corynebacterium species Issued by the Standards Unit, Microbiology Services, PHE Bacteriology – Identification | ID 2 | Issue no: 4.1 | Issue date: 29.10.14 | Page: 1 of 24 © Crown copyright 2014 Identification of Corynebacterium species Acknowledgments UK Standards for Microbiology Investigations (SMIs) are developed under the auspices of Public Health England (PHE) working in partnership with the National Health Service (NHS), Public Health Wales and with the professional organisations whose logos are displayed below and listed on the website https://www.gov.uk/uk- standards-for-microbiology-investigations-smi-quality-and-consistency-in-clinical- laboratories. SMIs are developed, reviewed and revised by various working groups which are overseen by a steering committee (see https://www.gov.uk/government/groups/standards-for-microbiology-investigations- steering-committee). The contributions of many individuals in clinical, specialist and reference laboratories who have provided information and comments during the development of this document are acknowledged. We are grateful to the Medical Editors for editing the medical content. For further information please contact us at: Standards Unit Microbiology Services Public Health England 61 Colindale Avenue London NW9 5EQ E-mail: [email protected] Website: https://www.gov.uk/uk-standards-for-microbiology-investigations-smi-quality- and-consistency-in-clinical-laboratories UK Standards for Microbiology Investigations are produced in association with: Logos correct at time of publishing. Bacteriology – Identification | ID 2 | Issue no: 4.1 | Issue date: 29.10.14 | Page: 2 of 24 UK Standards for Microbiology Investigations | Issued by the Standards Unit, Public Health England Identification of Corynebacterium species Contents ACKNOWLEDGMENTS ......................................................................................................... -

Azorhizobium Doebereinerae Sp. Nov
ARTICLE IN PRESS Systematic and Applied Microbiology 29 (2006) 197–206 www.elsevier.de/syapm Azorhizobium doebereinerae sp. Nov. Microsymbiont of Sesbania virgata (Caz.) Pers.$ Fa´tima Maria de Souza Moreiraa,Ã, Leonardo Cruzb,Se´rgio Miana de Fariac, Terence Marshd, Esperanza Martı´nez-Romeroe,Fa´bio de Oliveira Pedrosab, Rosa Maria Pitardc, J. Peter W. Youngf aDepto. Cieˆncia do solo, Universidade Federal de Lavras, C.P. 3037 , 37 200–000, Lavras, MG, Brazil bUniversidade Federal do Parana´, C.P. 19046, 81513-990, PR, Brazil cEmbrapa Agrobiologia, antiga estrada Rio, Sa˜o Paulo km 47, 23 851-970, Serope´dica, RJ, Brazil dCenter for Microbial Ecology, Michigan State University, MI 48824, USA eCentro de Investigacio´n sobre Fijacio´n de Nitro´geno, Universidad Nacional Auto´noma de Mexico, Apdo Postal 565-A, Cuernavaca, Mor, Me´xico fDepartment of Biology, University of York, PO Box 373, York YO10 5YW, UK Received 18 August 2005 Abstract Thirty-four rhizobium strains were isolated from root nodules of the fast-growing woody native species Sesbania virgata in different regions of southeast Brazil (Minas Gerais and Rio de Janeiro States). These isolates had cultural characteristics on YMA quite similar to Azorhizobium caulinodans (alkalinization, scant extracellular polysaccharide production, fast or intermediate growth rate). They exhibited a high similarity of phenotypic and genotypic characteristics among themselves and to a lesser extent with A. caulinodans. DNA:DNA hybridization and 16SrRNA sequences support their inclusion in the genus Azorhizobium, but not in the species A. caulinodans. The name A. doebereinerae is proposed, with isolate UFLA1-100 ( ¼ BR5401, ¼ LMG9993 ¼ SEMIA 6401) as the type strain. -

Corynebacterium Sp.|NML98-0116
1 Limnochorda_pilosa~GCF_001544015.1@NZ_AP014924=Bacteria-Firmicutes-Limnochordia-Limnochordales-Limnochordaceae-Limnochorda-Limnochorda_pilosa 0,9635 Ammonifex_degensii|KC4~GCF_000024605.1@NC_013385=Bacteria-Firmicutes-Clostridia-Thermoanaerobacterales-Thermoanaerobacteraceae-Ammonifex-Ammonifex_degensii 0,985 Symbiobacterium_thermophilum|IAM14863~GCF_000009905.1@NC_006177=Bacteria-Firmicutes-Clostridia-Clostridiales-Symbiobacteriaceae-Symbiobacterium-Symbiobacterium_thermophilum Varibaculum_timonense~GCF_900169515.1@NZ_LT827020=Bacteria-Actinobacteria-Actinobacteria-Actinomycetales-Actinomycetaceae-Varibaculum-Varibaculum_timonense 1 Rubrobacter_aplysinae~GCF_001029505.1@NZ_LEKH01000003=Bacteria-Actinobacteria-Rubrobacteria-Rubrobacterales-Rubrobacteraceae-Rubrobacter-Rubrobacter_aplysinae 0,975 Rubrobacter_xylanophilus|DSM9941~GCF_000014185.1@NC_008148=Bacteria-Actinobacteria-Rubrobacteria-Rubrobacterales-Rubrobacteraceae-Rubrobacter-Rubrobacter_xylanophilus 1 Rubrobacter_radiotolerans~GCF_000661895.1@NZ_CP007514=Bacteria-Actinobacteria-Rubrobacteria-Rubrobacterales-Rubrobacteraceae-Rubrobacter-Rubrobacter_radiotolerans Actinobacteria_bacterium_rbg_16_64_13~GCA_001768675.1@MELN01000053=Bacteria-Actinobacteria-unknown_class-unknown_order-unknown_family-unknown_genus-Actinobacteria_bacterium_rbg_16_64_13 1 Actinobacteria_bacterium_13_2_20cm_68_14~GCA_001914705.1@MNDB01000040=Bacteria-Actinobacteria-unknown_class-unknown_order-unknown_family-unknown_genus-Actinobacteria_bacterium_13_2_20cm_68_14 1 0,9803 Thermoleophilum_album~GCF_900108055.1@NZ_FNWJ01000001=Bacteria-Actinobacteria-Thermoleophilia-Thermoleophilales-Thermoleophilaceae-Thermoleophilum-Thermoleophilum_album -

Alpine Soil Bacterial Community and Environmental Filters Bahar Shahnavaz
Alpine soil bacterial community and environmental filters Bahar Shahnavaz To cite this version: Bahar Shahnavaz. Alpine soil bacterial community and environmental filters. Other [q-bio.OT]. Université Joseph-Fourier - Grenoble I, 2009. English. tel-00515414 HAL Id: tel-00515414 https://tel.archives-ouvertes.fr/tel-00515414 Submitted on 6 Sep 2010 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. THÈSE Pour l’obtention du titre de l'Université Joseph-Fourier - Grenoble 1 École Doctorale : Chimie et Sciences du Vivant Spécialité : Biodiversité, Écologie, Environnement Communautés bactériennes de sols alpins et filtres environnementaux Par Bahar SHAHNAVAZ Soutenue devant jury le 25 Septembre 2009 Composition du jury Dr. Thierry HEULIN Rapporteur Dr. Christian JEANTHON Rapporteur Dr. Sylvie NAZARET Examinateur Dr. Jean MARTIN Examinateur Dr. Yves JOUANNEAU Président du jury Dr. Roberto GEREMIA Directeur de thèse Thèse préparée au sien du Laboratoire d’Ecologie Alpine (LECA, UMR UJF- CNRS 5553) THÈSE Pour l’obtention du titre de Docteur de l’Université de Grenoble École Doctorale : Chimie et Sciences du Vivant Spécialité : Biodiversité, Écologie, Environnement Communautés bactériennes de sols alpins et filtres environnementaux Bahar SHAHNAVAZ Directeur : Roberto GEREMIA Soutenue devant jury le 25 Septembre 2009 Composition du jury Dr. -
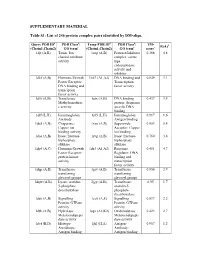
List of 246 Protein Complex Pairs Identified by DM-Align
SUPPLEMENTARY MATERIAL Table S1: List of 246 protein complex pairs identified by DM-align. Query PDB IDa PDB Classb: Temp PDB IDd PDB Classb: TM- R(Å)f (Chain1,Chain2) GO termc (Chain1,Chain2) GO termc scoree 1djt (A,B) Toxin: Ion 1aap (A,B) Protease/Inhibitor 0.368 4.8 channel inhibitor complex: serine activity type endopeptidase activity and inhibitor 1dkf (A,B) Hormone/Growth 1xb7 (A1,A2) DNA binding and 0.849 3.1 Factor Receptor: Transcription DNA binding and factor activity transciption factor activity 1dl5 (A,B) Transferase: 1utx (A,B) DNA binding 0.437 3.9 Methyltransferas protein: Sequence e activity specific DNA binding 1dlf (L,H) Immunoglobin: 1j05 (L,H) Immunoglobin: 0.917 1.6 Antibody Antigen binding 1do5 (A,B) Chaperone: 1xso (A,B) Superoxide 0.953 0.9 Copper ion Acceptor: Copper binding activity ion binding 1dos (A,B) lyase: fructose- 1rvg (A,B) lyase: fructose- 0.760 3.6 biphosphate biphosphate aldolase aldolase 1dp4 (A,C) Hormone/Growth 1dz3 (A1,A2) Response 0.481 4.7 Factor Receptor: Regulator: DNA protein kinase binding and activity transcription factor activity 1dqp (A,B) Transferase: 1grv (A,B) Transferase: 0.856 2.9 transferring transferring glycosyl groups glycosyl groups 1dqw (A,B) Lyase: orotidin- 2jgy (A,B) Transferase: 0.95 1.7 5-phosphate orotidin-5- decarboxylase phosphate decarboxylase 1ds6 (A,B) Signalling 1cc0 (A,E) Signalling 0.897 2.2 Protein: GTPase Protein: GTPase activity activity 1dth (A,B) Hydrolase: 1sqv (A2,K2) Oxidoredutase: 0.423 2.7 Metaloendopepti Metaloendopepti dase activity dase activity 1dvf -

Attachment 2 a Risk Profile of Dairy Products in Australia Appendices 1
2-06 22 March 2006 Attachment 2 A Risk Profile of Dairy Products in Australia Appendices 1-6 DRAFT ASSESSMENT REPORT PROPOSAL P296 PRIMARY PRODUCTION AND PROCESSING STANDARD FOR DAIRY RISK PROFILE OF DAIRY PRODUCTS IN AUSTRALIA VII APPENDICES 1. IMPACT OF PROCESSING ON DAIRY PRODUCT SAFETY 1.1 Milk and cream 1.2 Cheese 1.3 Dried milk powders 1.4 Infant formulae 1.5 Concentrated milk products 1.6 Butter and butter products 1.7 Ice cream 1.8 Cultured and fermented milk products 1.9 Dairy desserts 1.10 Dairy based dips 1.11 Casein, why products and other functional milk derivatives 1.12 Colostrum 2. EPIDEMIOLOGICAL INFORMATION ON OUTBREAKS OF FOODBORNE ILLNESS ASSOCIATED WITH DAIRY PRODUCTS 3. OCCURRENCE OF MICROBIOLOGICAL HAZARDS ASSOCIATED WITH DAIRY PRODUCTS 4. CONSUMPTION FIGURES OF DAIRY PRODUCTS FOR AUSTRALIAN CONSUMERS 5. HAZARD IDENTIFICATION/HAZARD CHARACTERISATION OF PATHOGENS 5.1 Aeromonas Spp. 5.2 Bacillus cereus 5.3 Brucella Spp. 5.4 Campylobacter jejuni/coli 5.5 Clostridium Spp. 5.6 Coxiella burnetii 5.7 Corynebacterium ulcerans 5.8 Cryptosporidium 5.9 Enterobacter sakazakii 5.10 Pathogenic Escherichia coli 5.11 Listeria monocytogenes 5.12 Mycobacterium bovis 5.13 Mycobacterium avium subsp. paratuberculosis 5.14 Salmonella Spp. 5.15 Shigella Spp. 5.16 Staphylococcus aureus 5.17 Streptococcus Spp. 5.18 Yersinia enterocolitica 6. PREVIOUS RISK ASSESSMENTS ON MICROBIOLOGICAL PATHOGENS IN DAIRY PRODUCTS 7. CHEMICAL RISK ASSESSMENT FRAMEWORK 8. REGULATORY FRAMEWORK FOR AGRICULTURAL AND VETERINARY CHEMICALS 9. MAXIMUM RESIDUE LIMITS 10. CHEMICAL RESIDUES MEASURED IN BOVINE DAIRY PRODUCTS 11. REGISTERED ANTIMICROBIAL AGENTS 12. -

Xanthobacter Flavus, a New Species of Nitrogen-Fixing Hydrogen Bacteria
INTERNATIONALJOURNAL OF SYSTEMATICBACTERIOLOGY, Oct. 1979, p. 283-287 Vol. 29, No. 4 0020-7713/79/04-0283/05$02.00/0 Xanthobacter flavus, a New Species of Nitrogen-Fixing Hydrogen Bacteria K. A. MALIK AND D. CLAUS Deutsche Sammlung von Mikroorganismen, Gesellschaft fur Biotechnologische Forschung mbH, D-3400 Gottingen, Federal Republic of Germany Mycobacterium flavum strain 301 (= Deutsche Sammlung von Mikroorganis- men 338), a hydrogen-utilizing bacterium, is capable of fixing molecular nitrogen and resembles other nitrogen-fixing hydrogen bacteria. However, it is clearly different in many characters from other strains of M. flavum (Orla-Jensen) Jensen (syn.: Micro bacterium flavum Orla-Jensen). It does resemble strains of Xantho- bacter Wiegel et al. with respect to cell wall composition, production of carotenoid pigments, carbon source utilization pattern, and deoxyribonucleic acid (DNA) base composition (69 mol% guanine + cytosine). Strain 301 is here regarded as belonging to a new and distinct species, for which the name Xanthobacter flauus is proposed. Strain 301 is the type strain of this species. X. flavus differs from Xanthobacter autotrophicus, the only other species in this genus to date, in several respects, and the DNA-DNA hybridization between X. flavus and X. autotrophicus is only 25%. In 1961 Federov and Kalininskaya (13) iso- Tsukamura (24) showed that unlike most rap- lated an aerobic, nitrogen-fixing bacterium idly growing mycobacteria, strain 301 has no (strain 301) from turf podzol soils and identified mycobactin. Mycobacteria are usually gram pos- it according to Krasil’nikov’s scheme (18) as a itive and strongly acid fast, but strain 301 is member of Mycobacterium flavum (Orla-Jen- gram variable and weakly acid fast. -

Using MALDI-TOF MS Analysis for Rapid Discrimination of MRSA MLST
Using MALDI-TOF MS analysis for rapid discrimination of MRSA MLST types Jang-Jih Lu1,2,Tsui-Ping Liu1, Shih-Cheng Chang1,2 1Department of Laboratory Medicine, Chang Gung Memorial Hospital, Linkou, Taoyuan, Taiwan 2Department of Medical Biotechnology and Laboratory Science, College of Medicine, Chang Gung University, Taoyuan, Taiwan Email: [email protected] 73 0:F8 MS Ra w 71 0:F9 MS Ra w 5000 6422.794 6000 Intens.[a.u.] Intens.[a.u.] Introduction 4000 Results ST5 ST5 4000 3000 2978.985 2000 6551.531 Staphylococcus aureus is one of the common causes of 3027.802 3054.785 2964.900 3175.943 2000 Table 1: MRSA and MSSA associated peaks by MALDI TOF 1000 community-associated infections and health care related 234 0:F1 MS Ra w 234 0:F1 MS Ra w 3005.919 MRSA(129 isolates) MSSA(77 isolates) 5000 6000 Intens.[a.u.] Intens.[a.u.] pathogen. It can usually cause wound infection, ST45 4000 ST45 6549.206 4000 3000 MS peaks (m/z) no. of isolate % of isolate no. of isolate % of isolate 6420.525 osteomyelitis, and even serious invasive infections, such as 2000 6610.628 2000 2978.373 3053.663 3174.805 2415 67 51.9 2 2.6 1000 sepsis and pneumonia. Epidemiology studies have shown 86 0:E4 MS Ra w 70 0:E5 MS Ra w 3058.565 5000 2431 65 50.4 0 0 3074.390 6000 Intens.[a.u.] Intens.[a.u.] that there are close relationship between the results of ST59 4000 6550.723 ST59 4000 3000 2879 51 39.5 2 2.6 6422.151 genotyping from multilocus sequence typing (MLST) and 2000 2000 characteristics of MRSA strains, including hospital- 6593 77 59.7 7 9.1 1000 200 0:E8 MS Ra w 38 0:E11 MS Ra w Appear of any above 103 79.8 11 14.3 2978.890 5000 6000 Intens.[a.u.] Intens.[a.u.] 6421.658 acquired, community-associated strain and even the 2965.568 ST239 4000 ST239 peaks 4000 3000 none of above peaks 6590.270 26 20.1 66 85.7 2937.113 3177.333 2916.384 antibiogram of the strain. -

NGS-Based Phylogeny of Diphtheria-Related Pathogenicity Factors in Different Corynebacterium Spp
Dangel et al. BMC Microbiology (2019) 19:28 https://doi.org/10.1186/s12866-019-1402-1 RESEARCHARTICLE Open Access NGS-based phylogeny of diphtheria-related pathogenicity factors in different Corynebacterium spp. implies species- specific virulence transmission Alexandra Dangel1* , Anja Berger1,2*, Regina Konrad1 and Andreas Sing1,2 Abstract Background: Diphtheria toxin (DT) is produced by toxigenic strains of the human pathogen Corynebacterium diphtheriae as well as zoonotic C. ulcerans and C. pseudotuberculosis. Toxigenic strains may cause severe respiratory diphtheria, myocarditis, neurological damage or cutaneous diphtheria. The DT encoding tox gene is located in a mobile genomic region and tox variability between C. diphtheriae and C. ulcerans has been postulated based on sequences of a few isolates. In contrast, species-specific sequence analysis of the diphtheria toxin repressor gene (dtxR), occurring both in toxigenic and non-toxigenic Corynebacterium species, has not been done yet. We used whole genome sequencing data from 91 toxigenic and 46 non-toxigenic isolates of different pathogenic Corynebacterium species of animal or human origin to elucidate differences in extracted DT, DtxR and tox-surrounding genetic elements by a phylogenetic analysis in a large sample set. Results: Sequences of both DT and DtxR, extracted from whole genome sequencing data, could be classified in four distinct, nearly species-specific clades, corresponding to C. diphtheriae, C. pseudotuberculosis, C. ulcerans and atypical C. ulcerans from a non-toxigenic toxin gene-bearing wildlife cluster. Average amino acid similarities were above 99% for DT and DtxR within the four groups, but lower between them. For DT, subgroups below species level could be identified, correlating with different tox-comprising mobile genetic elements. -

Probing the Biosynthetic Diversity of Actinobacteria 29-01-2018
BSc. A. Roeters - Probing the biosynthetic diversity of actinobacteria 29-01-2018 PROBING THE BIOSYNTHETIC DIVERSITY OF ACTINOBACTERIA MSc. Thesis by Arne Roeters, supervised by dr. MH Medema and JC Navarro Munoz PhD. Bioinformatics department Wageningen university. ABSTRACT certain plants. In these relationships the actinobacteria provide nitrogen to the plant and in The Actinobacteria are a large phylum of Gram- return they take some of the plants saccharide 1,2 positive bacteria of which we harvest many reserves . Maybe even more important and interesting about these bacteria, are their clinically useful natural products. A large portion of secondary metabolites that can be used for medical these clinically useful products are made by the purposes3. A large part of the clinically available largest genus within this phylum, called antibiotics come from Actinobacteria, and Streptomyces. These products are made by especially the largest genus Streptomyces. This biosynthetic gene clusters (BGCs), which are genus produces over two-third of the clinically physically clustered genes on the genome. To find useful natural antibiotics with its natural product 4 more of these natural compounds, genome mining biosynthetic gene clusters . Not nearly all-natural compounds have been found yet, meaning that has become one of the most important tools in there might still be many more useful compounds bioinformatics. This new technique has given rise that are made by the biosynthetic pathways of to programs like antiSMASH (Medema, et al., Actinobacteria. These biosynthetic pathways 2011). Programs like this have created new consist of genes that are physically clustered challenges due to the large amount of BGCs they together on the chromosome forming so called 5–7 mine, to narrow the search for new interesting biosynthetic gene clusters (BGCs) .