N-\Omega\-Cyanoalkyl\Aminophenols, and Their Preparation
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

The Enthalpy of Formation of Organic Compounds with “Chemical Accuracy”
chemengineering Article Group Contribution Revisited: The Enthalpy of Formation of Organic Compounds with “Chemical Accuracy” Robert J. Meier Pro-Deo Consultant, 52525 Heinsberg, North-Rhine Westphalia, Germany; [email protected] Abstract: Group contribution (GC) methods to predict thermochemical properties are of eminent importance to process design. Compared to previous works, we present an improved group contri- bution parametrization for the heat of formation of organic molecules exhibiting chemical accuracy, i.e., a maximum 1 kcal/mol (4.2 kJ/mol) difference between the experiment and model, while, at the same time, minimizing the number of parameters. The latter is extremely important as too many parameters lead to overfitting and, therewith, to more or less serious incorrect predictions for molecules that were not within the data set used for parametrization. Moreover, it was found to be important to explicitly account for common chemical knowledge, e.g., geminal effects or ring strain. The group-related parameters were determined step-wise: first, alkanes only, and then only one additional group in the next class of molecules. This ensures unique and optimal parameter values for each chemical group. All data will be made available, enabling other researchers to extend the set to other classes of molecules. Keywords: enthalpy of formation; thermodynamics; molecular modeling; group contribution method; quantum mechanical method; chemical accuracy; process design Citation: Meier, R.J. Group Contribution Revisited: The Enthalpy of Formation of Organic Compounds with “Chemical Accuracy”. 1. Introduction ChemEngineering 2021, 5, 24. To understand chemical reactivity and/or chemical equilibria, knowledge of thermo- o https://doi.org/10.3390/ dynamic properties such as gas-phase standard enthalpy of formation DfH gas is a necessity. -

Supporting Information
Photocatalytic Atom Transfer Radical Addition to Olefins utilizing Novel Photocatalysts Errika Voutyritsa, Ierasia Triandafillidi, Nikolaos V. Tzouras, Nikolaos F. Nikitas, Eleftherios K. Pefkianakis, Georgios C. Vougioukalakis* and Christoforos G. Kokotos* Laboratory of Organic Chemistry, Department of Chemistry, National and Kapodistrian University of Athens, Panepistimiopolis, Athens 15771, Greece SUPPORTING INFORMATION Page General Remarks S2 Optimization of the Reaction Conditions for the Photocatalytic Reaction S3 between 1-decene and BrCH2CN Synthesis of Photocatalysts S5 Synthesis of the Starting Materials S12 General Procedure for the Photocatalytic Reaction between Olefins and S18 BrCH2CN General Procedure for the Photocatalytic Reaction between Olefins and S29 BrCCl3 Determination of the Quantum Yield S33 Phosphorescence Quenching Studies S36 References S41 NMR Spectra S43 E. Voutyritsa, I. Triandafillidi, N. V. Tzouras, N. F. Nikitas, E. K. Pefkianakis, G. C. Vougioukalakis & C. G. Kokotos S1 General Remarks Chromatographic purification of products was accomplished using forced-flow ® chromatography on Merck Kieselgel 60 F254 230-400 mesh. Thin-layer chromatography (TLC) was performed on aluminum backed silica plates (0.2 mm, 60 F254). Visualization of the developed chromatogram was performed by fluorescence quenching, using phosphomolybdic acid, anisaldehyde or potassium permanganate stains. Mass spectra (ESI) were recorded on a Finningan® Surveyor MSQ LC-MS spectrometer. HRMS spectra were recorded on Bruker® Maxis Impact QTOF spectrometer. 1H and 13C NMR spectra were recorded on Varian® Mercury (200 MHz and 50 MHz respectively), or a Bruker® Avance (500 MHz and 125 MHz), and are internally referenced to residual solvent signals. Data for 1H NMR are reported as follows: chemical shift (δ ppm), integration, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, br s = broad singlet), coupling constant and assignment. -
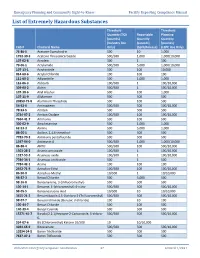
List of Extremely Hazardous Substances
Emergency Planning and Community Right-to-Know Facility Reporting Compliance Manual List of Extremely Hazardous Substances Threshold Threshold Quantity (TQ) Reportable Planning (pounds) Quantity Quantity (Industry Use (pounds) (pounds) CAS # Chemical Name Only) (Spill/Release) (LEPC Use Only) 75-86-5 Acetone Cyanohydrin 500 10 1,000 1752-30-3 Acetone Thiosemicarbazide 500/500 1,000 1,000/10,000 107-02-8 Acrolein 500 1 500 79-06-1 Acrylamide 500/500 5,000 1,000/10,000 107-13-1 Acrylonitrile 500 100 10,000 814-68-6 Acrylyl Chloride 100 100 100 111-69-3 Adiponitrile 500 1,000 1,000 116-06-3 Aldicarb 100/500 1 100/10,000 309-00-2 Aldrin 500/500 1 500/10,000 107-18-6 Allyl Alcohol 500 100 1,000 107-11-9 Allylamine 500 500 500 20859-73-8 Aluminum Phosphide 500 100 500 54-62-6 Aminopterin 500/500 500 500/10,000 78-53-5 Amiton 500 500 500 3734-97-2 Amiton Oxalate 100/500 100 100/10,000 7664-41-7 Ammonia 500 100 500 300-62-9 Amphetamine 500 1,000 1,000 62-53-3 Aniline 500 5,000 1,000 88-05-1 Aniline, 2,4,6-trimethyl- 500 500 500 7783-70-2 Antimony pentafluoride 500 500 500 1397-94-0 Antimycin A 500/500 1,000 1,000/10,000 86-88-4 ANTU 500/500 100 500/10,000 1303-28-2 Arsenic pentoxide 100/500 1 100/10,000 1327-53-3 Arsenous oxide 100/500 1 100/10,000 7784-34-1 Arsenous trichloride 500 1 500 7784-42-1 Arsine 100 100 100 2642-71-9 Azinphos-Ethyl 100/500 100 100/10,000 86-50-0 Azinphos-Methyl 10/500 1 10/10,000 98-87-3 Benzal Chloride 500 5,000 500 98-16-8 Benzenamine, 3-(trifluoromethyl)- 500 500 500 100-14-1 Benzene, 1-(chloromethyl)-4-nitro- 500/500 -

List of Lists
United States Office of Solid Waste EPA 550-B-10-001 Environmental Protection and Emergency Response May 2010 Agency www.epa.gov/emergencies LIST OF LISTS Consolidated List of Chemicals Subject to the Emergency Planning and Community Right- To-Know Act (EPCRA), Comprehensive Environmental Response, Compensation and Liability Act (CERCLA) and Section 112(r) of the Clean Air Act • EPCRA Section 302 Extremely Hazardous Substances • CERCLA Hazardous Substances • EPCRA Section 313 Toxic Chemicals • CAA 112(r) Regulated Chemicals For Accidental Release Prevention Office of Emergency Management This page intentionally left blank. TABLE OF CONTENTS Page Introduction................................................................................................................................................ i List of Lists – Conslidated List of Chemicals (by CAS #) Subject to the Emergency Planning and Community Right-to-Know Act (EPCRA), Comprehensive Environmental Response, Compensation and Liability Act (CERCLA) and Section 112(r) of the Clean Air Act ................................................. 1 Appendix A: Alphabetical Listing of Consolidated List ..................................................................... A-1 Appendix B: Radionuclides Listed Under CERCLA .......................................................................... B-1 Appendix C: RCRA Waste Streams and Unlisted Hazardous Wastes................................................ C-1 This page intentionally left blank. LIST OF LISTS Consolidated List of Chemicals -

Laboratory and Chemical Storage Areas Must Be Kept Locked When Not in Use
CHEMICAL HYGIENE PLAN AND HAZARDOUS MATERIALS SAFETY MANUAL FOR LABORATORIES This is the Chemical Hygiene Plan specific to the following areas: Laboratory name or room number(s): ___________________________________ Building: __________________________________________________________ Supervisor: _______________________________________________________ Department: _______________________________________________________ EMERGENCY TELEPHONE NUMBERS: 911 for Emergency and urgent consultation 3-2035 MSU Police; 3-2066 Facilities Management 3-2179 or (606) 207-0629 Eddie Frazier, Director of Risk Management 3-2584 or (606) 207-9425 Holly Niehoff, Environmental Health & Safety Specialist 3-2099 Derek Lewis, Environmental Health & Safety Technician 3-______ Building Supervisor State 24-hour warning point for HAZMAT Spill Notification If you need to report a spill in accordance with SARA Title III Section 304 and KRS 39E.190, please contact the Duty Officer at the Commonwealth Emergency Operations Center at 800.255.2587 which serves as the twenty-four (24) hour warning point and contact for the Commonwealth Emergency Response Commission. Updated 2018 All laboratory chemical use areas must maintain a work-area specific Chemical Hygiene Plan which conforms to the requirements of the OSHA Laboratory Standard 29 CFR 19190.1450. Morehead State UniversityMorehead laboratories State may Chemical use this document Hygiene Plan as a starting point for creating their work area specific Chemical Hygiene Plan (CHP). MSU Laboratory Awareness Certification For CHP of (Professor, Building, Room)________________________ The Occupational Safety and Health Administration (OSHA) requires that laboratory employees be made aware of the Chemical Hygiene Plan at their place of employment (29 CFR 1910.1450). The Morehead State University Chemical Hygiene Plan and Hazardous Materials Safety Manual serves as the written Chemical Hygiene Plan (CHP) for laboratories using chemicals at Morehead State University. -

Basics of Pump-And-Treat Ground-Water Remediation Technology
United States Robert S. Kerr EPA/600/8-90/003 Environmental Protection Environmental Research Laboratory March 1990 Agency Ada OK 74820 Research and Development Basics of Pump-and-Treat Ground-Water Remediation Technology Word-searchable version – Not a true copy EPA-600/8-90/003 Basics of Pump-and-Treat Ground-Water Remediation Technology James W. Mercer, David C. Skipp and Daniel Giffin Geo Trans, Inc. 250-A Exchange Place Herndon, Virginia 22070 Project Officer Randall R. Ross Extramural Activities and Assistance Division Robert S. Kerr Environmental Research Laboratory Office of Research and Development U.S. Environment Protection Agency Ada, Oklahoma 74820 Word-Searchable Version – Not a true copy Disclaimer The Information in this document has been funded in part by the United States Environmental Protection Agency under Contract No. 68-C8-0058 to Dynamac Corporation. It has been subjected to the Agency's peer and administrative review, and it has been approved for publication as an EPA document. Mention of trade names or commercial products does not constitute endorsement or recommendation for use. ii Word-searchable version – Not a true copy Foreword EPA is charged by Congress to protect the nation's land, air and water systems. Under a mandate of national environmental laws focused on air and water quality, solid waste management and the control of toxic substances, pesticides, noise and radiation, the Agency strives to formulate and implement actions which lead to a compatible balance between human activities and the ability of natural systems to support and nurture life. The Robert S. Kerr Environmental Research Laboratory is the Agency's center of expertise for investigation of the soil and subsurface environment. -

Environmental Protection Agency Pt. 355, App. B
Environmental Protection Agency Pt. 355, App. B [Alphabetical Order] Reportable Threshold plan- CAS No. Chemical name Notes quantity * ning quantity (pounds) (pounds) 5344–82–1 ............ Thiourea, (2-Chlorophenyl)- ....................................... ..................... 100 100/10,000 614–78–8 .............. Thiourea, (2-Methylphenyl)- ....................................... ..................... 500 500/10,000 7550–45–0 ............ Titanium Tetrachloride ................................................ ..................... 1,000 100 584–84–9 .............. Toluene 2,4-Diisocyanate ........................................... ..................... 100 500 91–08–7 ................ Toluene 2,6-Diisocyanate ........................................... ..................... 100 100 110–57–6 .............. Trans-1,4-Dichlorobutene ........................................... ..................... 500 500 1031–47–6 ............ Triamiphos .................................................................. ..................... 500 500/10,000 24017–47–8 .......... Triazofos ..................................................................... ..................... 500 500 76–02–8 ................ Trichloroacetyl Chloride .............................................. ..................... 500 500 115–21–9 .............. Trichloroethylsilane ..................................................... d .................. 500 500 327–98–0 .............. Trichloronate ............................................................... e ................. -

Chemicals Subject to TSCA Section 12(B) Export Notification Requirements (January 16, 2020)
Chemicals Subject to TSCA Section 12(b) Export Notification Requirements (January 16, 2020) All of the chemical substances appearing on this list are subject to the Toxic Substances Control Act (TSCA) section 12(b) export notification requirements delineated at 40 CFR part 707, subpart D. The chemicals in the following tables are listed under three (3) sections: Substances to be reported by Notification Name; Substances to be reported by Mixture and Notification Name; and Category Tables. TSCA Regulatory Actions Triggering Section 12(b) Export Notification TSCA section 12(b) requires any person who exports or intends to export a chemical substance or mixture to notify the Environmental Protection Agency (EPA) of such exportation if any of the following actions have been taken under TSCA with respect to that chemical substance or mixture: (1) data are required under section 4 or 5(b), (2) an order has been issued under section 5, (3) a rule has been proposed or promulgated under section 5 or 6, or (4) an action is pending, or relief has been granted under section 5 or 7. Other Section 12(b) Export Notification Considerations The following additional provisions are included in the Agency's regulations implementing section 12(b) of TSCA (i.e. 40 CFR part 707, subpart D): (a) No notice of export will be required for articles, except PCB articles, unless the Agency so requires in the context of individual section 5, 6, or 7 actions. (b) Any person who exports or intends to export polychlorinated biphenyls (PCBs) or PCB articles, for any purpose other than disposal, shall notify EPA of such intent or exportation under section 12(b). -

Environmental Protection Agency Pt. 355, App. A
Environmental Protection Agency Pt. 355, App. A Release means any spilling, leaking, the facility is located. In the absence pumping, pouring, emitting, emptying, of a SERC for a State or Indian Tribe, discharging, injecting, escaping, leach- the Governor or the chief executive of- ing, dumping, or disposing into the en- ficer of the tribe, respectively, shall be vironment (including the abandonment the SERC. Where there is a cooperative or discarding of barrels, containers, agreement between a State and a and other closed receptacles) of any Tribe, the SERC shall be the entity hazardous chemical, EHS, or CERCLA identified in the agreement. hazardous substance. Solution means any aqueous or or- Reportable quantity means, for any ganic solutions, slurries, viscous solu- CERCLA hazardous substance, the tions, suspensions, emulsions, or quantity established in Table 302.4 of 40 pastes. CFR 302.4, for such substance. For any State means any State of the United EHS, reportable quantity means the States, the District of Columbia, the quantity established in Appendices A Commonwealth of Puerto Rico, Guam, and B of this part for such substance. American Samoa, the United States Unless and until superseded by regula- Virgin Islands, the Northern Mariana tions establishing a reportable quan- Islands, any other territory or posses- tity for newly listed EHSs or CERCLA sion over which the United States has hazardous substances, a weight of 1 jurisdiction and Indian Country. pound shall be the reportable quantity. Threshold planning quantity means, SERC means the State Emergency for a substance listed in Appendices A Response Commission for the State in and B of this part, the quantity listed which the facility is located except in the column ‘‘threshold planning where the facility is located in Indian quantity’’ for that substance. -

Agrilife Hazcom Program
TEXAS AGRILIFE RESEARCH AND TEXAS AGRILIFE EXTENSION HAZARD COMMUNICATION PROGRAM Revised August 2010 Table of Contents INTRODUCTION: ..................................................................................................................................... 3 PROGRAM EXEMPTIONS AND EXCEPTIONS ................................................................................. 3 RESEARCH LABORATORY EXEMPTIONS ....................................................................................... 4 DUTIES AND RESPONSIBILITIES ........................................................................................................ 4 THE SAFETY COORDINATOR .................................................................................................................. 4 THE CENTER DIRECTOR ......................................................................................................................... 4 THE SAFETY OFFICER ............................................................................................................................. 5 SUPERVISORS ........................................................................................................................................... 5 EMPLOYEES ............................................................................................................................................. 5 CONTRACTED CONSTRUCTION, REPAIR AND MAINTENANCE ............................................................. 5 HAZARDOUS CHEMICAL INVENTORY: .......................................................................................... -

Anti-Inflammatory Agents
Europaisches Patentamt ® European Patent Office © Publication number: 0 276 065 Office europeen des brevets A1 © EUROPEAN PATENT APPLICATION © Application number: 88300163.8 © Int. CI.4: c 07 59/90 c 07 49/84, C 07( 59/92, © Date of filing: 11.01.88 c 07 65/40, C 07( 69/738, c 07 69/76, C 07( 93/06, c 07 103/178, c 07 103/58, c 07 117/00, C 07 C 121/34 © Priority: 12.01.87 US 2542 © Applicant: ELI LILLY AND COMPANY Lilly Corporate Center © Date of publication of application: Indianapolis Indiana 46285 (US) 27.07.88 Bulletin 88/30 @ Inventor: Bollinger, Nancy Grace Designated Contracting States: 7601 East 80th Street AT BE CH DE ES FR GB GR IT LI LU NL SE Indianapolis Indiana 46256 (US) Goodson, Theodore, Jr. i 4045 Devon Drive Indianapolis Indiana 46226 (US) Herron, David Kent 5945 Andover Road Indianapolis Indiana 46220 (US) @ Representative: Tapping, Kenneth George et al Erl Wood Manor Windlesham Surrey, GU20 6PH (GB) A request for correction the originally filed description pages 2, 3, 4, 8, 14, and 50 and claim 1 has been filed pursuant to Rule 88 EPC. A decision on the request will be taken during the proceedings before the Examining Division (Guidelines for Examination in the EPO, A-V, 2.2). © Anti-inflammatory agents. © This invention provides benzene derivatives of the Formula > Ri-Z- > 0-A-R4 (O o CO pharmaceutical formulations of those derivatives, and a method is. of using the derivatives for the treatment of inflammation in CM mammals. -

1 Titan's Organic Aerosols: Molecular Composition And
TITAN’S ORGANIC AEROSOLS: MOLECULAR COMPOSITION AND STRUCTURE OF LABORATORY ANALOGUES INFERRED FROM PYROLYSIS GAS CHROMATOGRAPHY MASS SPECTROMETRY ANALYSIS Marietta Morissona,b,*, Cyril Szopab,c, NatHalie Carrascob,c, Arnaud BucHa and Thomas Gautierd a LGPM Laboratoire de Génie des Procédés et Matériaux, Ecole CentraleSupelec, Châtenay-Malabry Cedex, France b LATMOS/IPSL, UVSQ Université Paris-Saclay, UPMC Univ. Paris 06, CNRS, Guyancourt, France c Institut Universitaire de France, Paris F-75005, France d NASA Goddard Space FligHt Center, Greenbelt, MD, United States Abstract Analogues of Titan’s aerosols are of primary interest in the understanding of Titan’s atmospheric chemistry and climate, and in tHe development of in situ instrumentation for future space missions. Numerous studies have been carried out to characterize laboratory analogues of Titan aerosols (tholins), but their molecular composition and structure are still poorly known. If pyrolysis gas chromatograpHy mass spectrometry (pyr-GCMS) Has been used for years to give clues about their chemical composition, HigHly disparate results were obtained witH tHis tecHnique. THey can be attributed to tHe variety of analytical conditions used for pyr-GCMS analyses, and/or to differences in the nature of the analogues analyzed, tHat were produced witH different laboratory set-ups under various operating conditions. In order to Have a better description of Titan’s tHolin’s molecular composition by pyr-GCMS, we carried out a systematic study witH two major objectives: (i) exploring tHe pyr-GCMS analytical parameters to find the optimal ones for the detection of a wide range of chemical products allowing a cHaracterization of tHe tHolins composition as comprehensive as possible, and (ii) HigHligHting tHe role of the CH4 ratio in tHe gaseous reactive medium on the tholin’s molecular structure.