Thesis-1971D-R917f.Pdf (2.777Mb)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

List of Legumes
Healthy Oils & SmokePoints When it comes to the cooking oil in your cupboard, there is a huge difference in healthfulness depending on how the oil is stored and how it will be used. First let’s get some definitions for commonly used terms on labels and discussions about oils. Term Definition Cold Pressed Extracted without using heat. Extracted using a screw-type machine that presses the oil out. Can be done Expeller Pressed slowly, with very little heat, or can be done quickly with lots of friction and high temperatures. The first cold pressing which contains the best tasting and most healthful oils. Must contain less than 1% acids. By definition, this is cold-pressed and first Extra Virgin pressed, so don’t need to see these terms on the label. Must say 100% extra virgin, or may be a blend. The first cold pressing, but can contain a little more acids than the extra-virgin Virgin (1-3 percent). Seeds that have been genetically manipulated to decrease the amount of High Oleic essential fatty acids so that they have a longer shelf life. Are left in their state after pressing – no filtering. These oils tend to be more Unrefined Oils flavorful and richer in nutrients, however they have a very low smoke point. Oils have their impurities filtered out, to increase stability and allow for higher Refined temperature cooking. The processing can use toxic solvents, caustic soda, bleaches and phosphoric acid. Smoke Point Stage at which a heated oil begins to smoke, just before it bursts into flames. HEALTHY OILS & SMOKE POINTS PAGE | 1 © 2021 Health-Naturally.org Term Definition Oil should smell and taste like the food it came from. -

Rabbi Eliyahu Shuman Director of Supervision Effective Through 11
June 2, 2021 22 Sivan 5781 La Tourangelle, Inc. 2340 East Main Street Suite 400 Woodland, CA 95776 This is to certify that the 39 products specified in the listing below, manufactured by LA TOURANGELLE, INC. of the above address, are Kosher and under our supervision. PLEASE NOTE THE FOLLOWING CONDITIONS OF CERTIFICATION: All products listed below are Pareve. All products listed below are certified only when bearing the Star-K symbol. All products listed below are Kosher for year-round use, excluding Passover. This letter of certification is valid through November 30, 2021 and is subject to renewal at that time. BRAND: La Tourangelle UKD# PRODUCT LISTING SK0HZG4N550 All Purpose Baking Spray SKDZZE3SG6W Avocado Oil SKVLKXREYAU Avocado Oil Non-Stick Cooking Spray SKB0NQF2TZU AvoCoco Oil SK5KPHXJ7WR Everyday Avocado Oil SK27270H2EA Expeller Pressed Vegetable Oil SK3YJF27B06 Grape Seed Oil SKZR5EBNPJE Grapeseed Oil Non-Stick Cooking Spray SKTA059CQ2D Organic Canola Oil SKEVY33L343 Organic Canola Oil Non-Stick Cooking Spray SKK08UW6IKZ Organic Expeller Pressed & Refined Coconut Oil SKAC2BUNNSR Organic Extra Virgin Coconut Oil SKN6ZPNS4QU Organic Extra Virgin Olive Oil SKIVAMH6Y1J Organic Extra Virgin Olive Oil Non-Stick Cooking Spray SKYKMZA08PT Organic Flaxseed Oil SKSDDTU4VXP Organic Refined Coconut Oil SK8C58TF7N1 Organic Sun Coco Oil SKGNEZ4A2KZ Organic Sunflower Oil SKR3YHL7L04 Organic Toasted Sunflower Oil SKWVAF83EW0 Organic Virgin Unrefined Coconut Oil SKQ4DM4KBR7 Pan Asian Stir Fry Oil Rabbi Eliyahu Shuman Effective Through 11/30/2021 Director of Supervision Page 1 of 2 June 2, 2021 22 Sivan 5781 La Tourangelle, Inc. 2340 East Main Street Suite 400 Woodland, CA 95776 PLEASE NOTE THE FOLLOWING CONDITIONS OF CERTIFICATION: All products listed below are Pareve. -

NPSA 75Th Anniversary Digital Cookbook
75TH ANNIVERSARY COOKBOOK WWW.ILOVEPECANS.ORG 75TH ANNIVERSARY COOKBOOK Celebrating 75 Ways to Enjoy Pecans With their crisp texture and rich, buttery flavor, pecans are remarkably versatile. To celebrate their many uses, we are bringing pecans to life with 75 amazing recipes in honor of the National Pecan Shellers Association’s 75th anniversary. Whether you’re a novice in the kitchen or an expert culinarian, you’ll find an abundance of delicious recipe choices. From new creations to traditional favorites, you’ll find a range of original ideas from savory to sweet and simple to complex. Each recipe provides easy-to-follow steps for preparing delicious and healthy snacks, appetizers, soups salads, entrees, side dishes, desserts, beverages and breakfast options. Explore the variety of original recipes created by nationally renowned chefs from companies like Baskin Robbins, Cinnabon, and Outback Steakhouse, as well as bloggers like SouthernBite. com. These culinary experts met at the test kitchens of publishing giant Time Inc. Food Studios to test and develop recipes that are easy to prepare and exciting to taste. Pecans - Beyond the Pie often less expensive than whole pecans – so purchase the chopped version and you’ll also Long a Southern tradition, more pecan pies save yourself an extra step in your preparation. are consumed in the United States between the Thanksgiving and Christmas holidays than any other time of year. That’s also when pecans Storing Pecans are at their best. But we have exciting recipes How you store your pecans will make a difference to enjoy pecans all year long like salads, in their freshness and flavor. -

Specialty Oils Is Preventing That Remains After Pressing, and Virgin Olive Oxidation―Especially in Those That Contain a High Level of Oil
[Fats/Oils] Vol. 18 No. 9 September 2008 Pouring on Specialty Oil Ideas By Tamie Cook, Contributing Editor They say variety is the spice of life. And, if that’s the case, the product and menu developer’s pantry can be a veritable smorgasbord of piquancy. Literally, we have the world at the end of our whisk, and this has never been truer than when it comes to reaching for oils. In days past, choices were largely limited to vegetable, peanut, soybean, the occasional variety of olive oil and, on a good day, extra virgin olive oil. No longer confined to these basics, myriad choices now address today’s ingredient requirements for health, variety, taste and versatility. Whether it’s for a new dish on the menu or the next big product line, look at the choices, learn about their many uses and try some new oil on your palette. Most fruit, nut and seed oils generally undergo comparatively little processing. Many are mechanically, or “expeller,” extracted and are unrefined so no solvent was used to extract the oil, and no bleach or deodorizing agent was used to refine it. Thus, the oil retains its full, natural flavor, aroma and color, and many of its naturally occurring nutrients. This translates into a product that not only adds flavor to a dish, but also adds value to a menu or product. Shedding new light on an old friend When it comes to olive oil, everything old is new again. The olive tree, a native of Asia Minor, has been around for more than 6,000 years. -
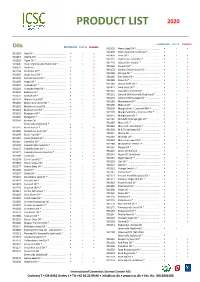
Product List 2020
PRODUCT LIST 2020 CONVENTIONAL ORGANIC STABILIZED Oils CONVENTIONAL ORGANIC STABILIZED 901199 Hemp Seed Oil * .................................. • • • 901499 Hemp Seed Oil Unrefined * ................. • • 901193 Acai Oil * ............................................. • • • 901450 Inchi Oil *............................................. • • • 901367 Alfalfa Oil* ........................................... • • • 901112 Jojoba Oil – Colorless * ........................ • • • 901228 Algae Oil * ........................................... • • • 901110 Jojoba Oil – Golden * ........................... • • • 907440 Aloe Oil (Internally Stabilized)* .......... • • 901162 Kakadu Oil * ........................................ • • • 906221 Amla Oil .............................................. • • 901152 Kalahari Melon Seed Oil * ................... • • • 901148 Andiroba Oil * ..................................... • • • 901168 Karanja Oil * ........................................ • • • 901387 Apple Seed Oil * .................................. • • • 901165 Kiwi Seed Oil * ..................................... • • • 901176 Apricot Kernel Oil * ............................. • • • 901185 Kukui Oil * ........................................... • • • 901195 Argan Oil * ........................................... • • • 901180 Lemon Seed Oil * ................................ • • • 901118 Avocado Oil * ...................................... • • • 901421 Lime Seed Oil * .................................... • • • 901218 Avocado Seed Oil * ............................. -

A-Anions 221 Acetate 23, 106 Acetate-Malonate Pathway 24
Index a-anions 221 anti-block agent 218, 237 acetate 23, 106 antioxidants 84, 173, 175, 176 acetate-malonate pathway 24 anti-slip agent 218, 237 acetylenic acids 10, 11, 159 Apis mellifera 85 acetylenic intermediates 30 apricot oil 66, 67 acid anhydrides 214 arachidic acid 5, 6 acid chlorides 214 arachidonic acid 8,9, 29, 184 acidity 102 synthesis 31, 32 acidolysis 208,209, 213 Arachis hypogaea 67 ACP, see acyl carrier protein argentation tic 19 activated carbon 90 arsenites 18 acyl carrier protein 25 asci epic acid 7 acylation 47 Aspergillus niger 88, 97, 213 acylglycerols 36, 52 Aspergillus oryzae 213 acylpyrrolidide 154 auricolic acid 13, 14 alchornoic acid 15 autoxidation 162-167 alcohols 217,238 avenasterol 84 alcoholysis 208, 209 avocado oil 67 alepramic acid 14 aziridines 202 aleprestic acid 14 alepric acid 14 babassu oil 88 aleprolic acid 14 beef tallow 72 aleprylic acid 14 beeswax 84 Aleurites fordii 10 behenic acid 5, 6 alkanoic acids, crystal structure 130 bentonitite 90 alkylbenzoxazole 154 benzyl-sn-glycerol 54 alkyl dimethyloxazole 154 benzylidene glycerol 48 alkyl ketene dimers (AKD) 215 Betapol 55, 214 alkyl polyglucoside 217 BHA, see butylated hydroxyanisole allenic acids 10, 11, 39 BHT, see butylated hydroxy toluene allenic epoxides 187 biodiesel 241 allylic bromination 194 biohydrogenation 161 alklylic hydroperoxides 162, 163, 165 biosynthesis 24, 44 almond oil 66, 67, 69 blackcurrant oil 64, 65 alternation 130 bleaching 89, 90 amides 218, 238 Bligh and Dyer method 101 amine oxides 219, 220, 237 BOB, see oleodibehenin -

SOME of the MINOR OIL CROPS 593 One Another in Composition And, In- Two, Crushing the Kernel Into Fragments Deed, Are Not Readily Difterentiated
Some of the Minor Oil importations. As recoverable oils from domestic crops, they are still in the class of minor oils. It must be borne in mind that the Crops existence of a. potential supply of any given oil does not mean that a profit can be realized by recovering it, even Ernest B. Rester at high market prices. Numerous eco- nomic factors have to be considered. By oils and fats, w^e usually mean the nonv^olatile oils that occur naturally as In ordinary times we could get along chemical combinations of glycerin and very well with an adequate supply of fatty acids. We shall not consider here the oils and fats from soybeans, pea- the volatile or essential oils that are nuts, cottonseed, corn, and milk^ which valued for their fragrance or flavor, we produce or import in huge amounts. such as peppermint, wintergreen, and But in times of emergency, when out- citronella. Also excluded are certain side supplies are cut off, we must sup- vegetable waxe^s, which in many re- plement our domestic production with spects resemble oils and fats but do not every available pound from every have the glyceride structure. available source. That^ plus the fact Vegetable oils differ from one an- that some oils have properties that other mainly in the kind and amount adapt them to specific uses, is why we of combined fatty acids, and the classes, are interested in minor oil crops. kinds, and amounts of impurities pres- Furthermore, many of the minor ent. Therefore, the properties of the crops can be exploited as small-business various vegetable oils usually differ ventures and may yield a fair return to only in degree, except in a highly in- a few investors who care to go into the dividual oil that contains a large per- vegetable-oil industry with a moderate centage of some unique constituent. -

Feasibility Assessment of Investing in a Pecan Oil and Flour Processing
Feasibility Assessment of Investing in a Pecan Oil and Flour Processing Facility Using New Extraction Technology Research Report 778 Skyla Cockerham, William Gorman, Niels Maness, and Jay Lillywhite1 Agricultural Experiment Station • College of Agricultural, Consumer and Environmental Sciences ABSTRACT (Pirovano, 2010). They are also buying more specialty This study examines the feasibility of locating and oper- gourmet foods, particularly coffee, chocolate, specialty ating a 2-million-pound-capacity pecan processing facil- oils, cheese, and soft drinks (Wolfe, 2010). Sales of ity using a new extraction technology in southern New gluten-free products rose by 16% from 2009 to 2010 Mexico. The extraction technology is called the Okla- (Pirovano, 2010). These trends provide an opportunity homa State University/ATEP process, which involves to position pecan oil and flour in expanding markets. propane solvent extraction to produce two products: pecan oil and a de-fatted gluten-free pecan flour. We completed an in-depth market analysis, consumer prod- METHODS OF PRODUCING PECAN OIL uct research, financial analysis, and sensitivity analysis. AND FLOUR This project appears to be financially feasible, with a There are several techniques for extracting oil from pe- projected internal rate of return (IRR) of 18% over a cans and similar nuts. The three that were considered 20-year project life based on paying $4.22 per pound were 1) mechanical extrusion, 2) traditional solvent for pecans. However, the price of pecans increased by extraction methods, and 3) a new solvent extraction nearly 40% in 2010 due to a large increase in exports of method involving propane, referred to as the Oklahoma pecans to China. -

Factors Influencing Storage Quality of Hazelnut Varieties
AN ABSTRACT OF THE THESIS Kais S. Ebrahem for the degree of Doctor of philosophy in Horticulture presented on October 16. 1992 Title: FACTORS INFLUENCING STORAGE QUALITY OF HAZELNUT VARIETIES Abstract approved: . ^. Dr. Daryl G.\. RichaRichardson, Professor of Horticulture This thesis research is a series of five studies dealing with various aspects of hazelnut quality: 1. Identification of kernel mold and its incidence among hazelnut varieties. 2. Seasonal development and composition of kernels. 3. Hazelnut oil composition (fatty acids and tocopherols) of seventeen varieties. 4. Hazelnut oil composition compared to 14 other nuts and oilseeds. 5. Storage and roasting effects on lipid oxidation. Barcelona, Daviana, and Ennis varieties of hazelnuts used to study white mold showed that mold incidence was highest in 1986, and much less in 1987 and 1988. Ramularia spp was the only fungus that was identified in all samples with kernel mold and was usually found at high percentages. Temperatures for drying, storage, and numbers of nuts per cluster had no significant effect on percent mold. The second part of this study measured fatty acid and vitamin E concentrations during the growing season. Samples were collected from seven varieties (Barcelona, Daviana, Ennis, Tonda Romana, Tonda Gentile della Langhe, Tombul, and Tombul Ghiaghli). Kernel oil and vitamin E (tocopherol) concentrations increased with time, while moisture decreased. Oleic acid was the major fatty acid (ca.75%) found in hazelnuts, followed by linoleic(ca.20%). Linolenic was high at the beginning of the season but then decreased to about 1% at harvest. Alpha-tocopherol was the major form of vitamin E found, composing almost 95% of total tocopherols. -

Fatty Acid Composition and Its Relationship with Physicochemical Properties of Pecan (Carya Illinoensis) Oil J.F
Fatty Acid Composition and Its Relationship with Physicochemical Properties of Pecan (Carya illinoensis) Oil J.F. Toro-Vazquez*, M.A. Charó-Alonso, and F. Pérez-Briceño aFacultad de Ciencias Quémicas-CIEP, Universidad Autónoma de San Luis Potosí, San Luis Potosí 78210, México ABSTRACT: The composition and physicochemical proper- do so. However, it is important to point out that the results of ties of pecan (Carya illinoensis) kernels and oils from different Mattson and Grundy (1) have not been consistently demon- native trees of the central region of Mexico were investigated. strated by other investigators. Some of the problems that The main compositional characteristic of the kernel was the might be associated with this inconsistency of results have high lipid content (70–79% w/w on dry basis) with elevated been pointed out (2,3). The HDL are the lipoproteins respon- concentration of oleic acid (55–75% w/w). The results con- sible for transporting cholesterol to the liver where it is catab- firmed the relationship in the biosynthesis of linoleic and olized. Then, a reduction in the blood levels of HDL is not linolenic acids from oleic acid existing in oilseeds. Our results indicate that in pecans such relationship is a function of pecan advisable. The so-called Mediterranean diet, with a lipid tree age. The proportion of oleic, linoleic, and linolenic fatty source mainly based on olive oil (55–83% oleic acid), ob- acids determined the oxidative stability, viscosity, and melt- tained considerable attention, supported by additional claims ing/crystallization behavior of pecan oil. In general, these prop- of lower colon cancer mortality rates and lower incidence of erties in pecan oils were similar or superior to extra-virgin olive breast cancer in countries that follow this diet (e.g., Spain, oil and unrefined sesame oil. -

Pecans: a Healthful
COLLEGE OF AGRICULTURAL, CONSUMER AND ENVIRONMENTAL SCIENCES Pecans: A Healthful New Mexico-grown Food Revised by Richard Heerema, Raquel Garzon, and John Hartley1 aces.nmsu.edu/pubs • Cooperative Extension Service • Guide E-138 Did you know The College of that pecan nuts are the only ma- Agricultural, jor tree nut crop that originated in North America? Consumer and For centuries, pecan nuts were Environmental enjoyed by Na- tive Americans Sciences is an across what is now the south- central region of engine for economic the United States. | Dreamstime.com Makela © Mona American settlers and community in the 1800s brought pecan trees westward to New Mexico, and soon after recognized the Mesilla Valley’s soils and climate as ideally suited to produc- development in New ing top-quality pecans. As a result, southern New Mexico is now one of the world’s premier pecan growing areas, and pecan nuts have come to be fea- tured prominently in New Mexico’s distinctive cuisine. Mexico, improving Recently, pecans have received a lot of very positive attention because of a significant shift in how scientists now think about fats in the diet. We now the lives of New know that the fat in nuts and other foods has many positive health benefits. It wasn’t too long ago that people were discouraged from eating nuts due Mexicans through to their high fat content. Research, some of which was conducted at New Mexico State University, has shown that regular pecan nut consumption may in fact have a number of important health benefits, and recent studies have academic, research, shown that the pruning and fertilizer practices used by New Mexico farmers may actually enhance the health-promoting components of pecan kernels and Extension even more2! programs. -

Chemical Properties and Lipid Profile of Egyptian Pecan Nahid M
Assiut J. of Agric. Sci., 42 No.2 (222-237) Chemical Properties and Lipid Profile of Egyptian Pecan Nahid M. El-Shimy, Laila A. Shekib, Nesrin A. El-Shehy Food Science and Technology Department, Faculty of Agriculture, Alexandria University, Egypt. Abstract: linoleic and α-linolenic acids. Pecan is a promising crop The ratio between unsaturated that can replace walnuts and it fatty acids to saturated fatty acids can be consumed for its potential was 11.20:1 and 09.96:1 for De- health benefit. In this study two sirable and Moneymaker culti- cultivars of pecan (Carya illino- vars, respectively. Pecan kernels inensis) grown in Egypt; Desir- contained omega-3 fatty acids able and Moneymaker cultivars, (ώ3) family: α-linolenic acid, were investigated for their kernel eicosapentaenoic acid (EPA) and colours' chemical composition docosahexaenoic acid (DHA) in (moisture, sugars, starch, crude both cultivars. lipids , crude proteins, crude fi- Peroxide, p-anisidine, total bers and ash. Lipid oxidation of oxidation values (totox value), pecan was evaluated by measur- acidity, free fatty acids, and thio- ing: peroxide, p-anisidine, total barbituric acid value (TBA) of oxidation values and thiobarbi- Desirable cultivar were statisti- turic acid value was also deter- cally significant lower than those mined. Moreover, determination of Moneymaker cultivar. of fatty acid profile and minerals Keywords: Pecan (Carya illino- content were carried out. inensis), acidity value, free fatty The major chemical compo- acids value, peroxide value, p- sition in pecan cultivars was anisidine value, omega-3 fatty crude lipids (74.84% - 75.94%). acids, thiobarbituric acid value. Moneymaker had higher total Introduction : carbohydrates and sugars and Pecan (Carya illinoinensis), lower protein than those of De- which belongs to the family Jug- sirable cultivar, while no signifi- landaceae, is one of the oldest cant differences were observed in tree nuts in the world.