Development of Analytical Methods to Characterize Organic Aerosol and Black Carbon
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Alteration of Volatile Chemical Composition in Tobacco Plants Due to Green Peach Aphid (Myzus Persicae Sulzer) (Hemiptera: Aphididae) Feeding
Song et al.: Green peach aphid (Myzus persicae Sulzer) (Hemiptera: Aphididae) feeding alters volatile composition in tobacco plants - 159 - ALTERATION OF VOLATILE CHEMICAL COMPOSITION IN TOBACCO PLANTS DUE TO GREEN PEACH APHID (MYZUS PERSICAE SULZER) (HEMIPTERA: APHIDIDAE) FEEDING SONG, Y. Z.1,2,3 – GUO, Y. Q.1,2 – CAI, P. M.2,3 – CHEN, W. B.1,2 – LIU, C. M.1,2* 1Biological Control Research Institute, College of Plant Protection, Fujian Agriculture and Forestry University, Fuzhou 350002, China e-mail: [email protected] (Song, Y. Z.); [email protected] (Guo, Y. Q.); [email protected] (Chen, W. B.) 2State Key Laboratory of Ecological Pest Control for Fujian and Taiwan Crops, Fuzhou 350002, China e-mail: [email protected] (Cai, P. M.) 3Department of Horticulture, College of Tea and Food Science, Wuyi University, Wuyishan 354300, China *Corresponding author e-mail: [email protected]; phone: +86-0591-8378-9420; fax: +86-0591-8378-9421 (Received 10th Jul 2020; accepted 6th Oct 2020) Abstract. In response to insect pest herbivory, plants can generate volatile components that may serve multiple roles as communication signals and defence agents in a multitrophic context. In the present study, the volatile profiles of tobacco plants Nicotiana tabacum L., with and without infestation by sap-sucking aphids Myzus persicae Sulzer, were measured by gas chromatography-mass spectrometry (GC-MS). The results revealed that a total of 10 compounds were identified from healthy tobacco plants, and a total of 14 and 16 compounds were isolated from aphid-infested tobacco plants at 24 and 48 hours after infestation, respectively. -

Synthetic Turf Scientific Advisory Panel Meeting Materials
California Environmental Protection Agency Office of Environmental Health Hazard Assessment Synthetic Turf Study Synthetic Turf Scientific Advisory Panel Meeting May 31, 2019 MEETING MATERIALS THIS PAGE LEFT BLANK INTENTIONALLY Office of Environmental Health Hazard Assessment California Environmental Protection Agency Agenda Synthetic Turf Scientific Advisory Panel Meeting May 31, 2019, 9:30 a.m. – 4:00 p.m. 1001 I Street, CalEPA Headquarters Building, Sacramento Byron Sher Auditorium The agenda for this meeting is given below. The order of items on the agenda is provided for general reference only. The order in which items are taken up by the Panel is subject to change. 1. Welcome and Opening Remarks 2. Synthetic Turf and Playground Studies Overview 4. Synthetic Turf Field Exposure Model Exposure Equations Exposure Parameters 3. Non-Targeted Chemical Analysis Volatile Organics on Synthetic Turf Fields Non-Polar Organics Constituents in Crumb Rubber Polar Organic Constituents in Crumb Rubber 5. Public Comments: For members of the public attending in-person: Comments will be limited to three minutes per commenter. For members of the public attending via the internet: Comments may be sent via email to [email protected]. Email comments will be read aloud, up to three minutes each, by staff of OEHHA during the public comment period, as time allows. 6. Further Panel Discussion and Closing Remarks 7. Wrap Up and Adjournment Agenda Synthetic Turf Advisory Panel Meeting May 31, 2019 THIS PAGE LEFT BLANK INTENTIONALLY Office of Environmental Health Hazard Assessment California Environmental Protection Agency DRAFT for Discussion at May 2019 SAP Meeting. Table of Contents Synthetic Turf and Playground Studies Overview May 2019 Update ..... -

Vapor Pressures and Vaporization Enthalpies of the N-Alkanes from C31 to C38 at T ) 298.15 K by Correlation Gas Chromatography
BATCH: je3a29 USER: ckt69 DIV: @xyv04/data1/CLS_pj/GRP_je/JOB_i03/DIV_je030236t DATE: March 15, 2004 1 Vapor Pressures and Vaporization Enthalpies of the n-Alkanes from 2 C31 to C38 at T ) 298.15 K by Correlation Gas Chromatography 3 James S. Chickos* and William Hanshaw 4 Department of Chemistry and Biochemistry, University of MissourisSt. Louis, St. Louis, Missouri 63121, USA 5 6 The temperature dependence of gas-chromatographic retention times for henitriacontane to octatriacontane 7 is reported. These data are used in combination with other literature values to evaluate the vaporization 8 enthalpies and vapor pressures of these n-alkanes from T ) 298.15 to 575 K. The vapor pressure and 9 vaporization enthalpy results obtained are compared with existing literature data where possible and 10 found to be internally consistent. The vaporization enthalpies and vapor pressures obtained for the 11 n-alkanes are also tested directly and through the use of thermochemical cycles using literature values 12 for several polyaromatic hydrocarbons. Sublimation enthalpies are also calculated for hentriacontane to 13 octatriacontane by combining vaporization enthalpies with temperature adjusted fusion enthalpies. 14 15 Introduction vaporization enthalpies of C21 to C30 to include C31 to C38. 54 The vapor pressures and vaporization enthalpies of these 55 16 Polycyclic aromatic hydrocarbons (PAHs) are important larger n-alkanes have been evaluated using the technique 56 17 environmental contaminants, many of which exhibit very of correlation gas chromatography. This technique relies 57 18 low vapor pressures. In the gas phase, they exist mainly entirely on the use of standards in assessment. Standards 58 19 sorbed to aerosol particles. -
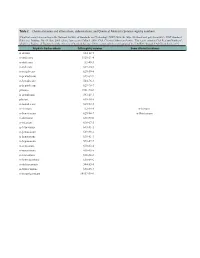
Table 2. Chemical Names and Alternatives, Abbreviations, and Chemical Abstracts Service Registry Numbers
Table 2. Chemical names and alternatives, abbreviations, and Chemical Abstracts Service registry numbers. [Final list compiled according to the National Institute of Standards and Technology (NIST) Web site (http://webbook.nist.gov/chemistry/); NIST Standard Reference Database No. 69, June 2005 release, last accessed May 9, 2008. CAS, Chemical Abstracts Service. This report contains CAS Registry Numbers®, which is a Registered Trademark of the American Chemical Society. CAS recommends the verification of the CASRNs through CAS Client ServicesSM] Aliphatic hydrocarbons CAS registry number Some alternative names n-decane 124-18-5 n-undecane 1120-21-4 n-dodecane 112-40-3 n-tridecane 629-50-5 n-tetradecane 629-59-4 n-pentadecane 629-62-9 n-hexadecane 544-76-3 n-heptadecane 629-78-7 pristane 1921-70-6 n-octadecane 593-45-3 phytane 638-36-8 n-nonadecane 629-92-5 n-eicosane 112-95-8 n-Icosane n-heneicosane 629-94-7 n-Henicosane n-docosane 629-97-0 n-tricosane 638-67-5 n-tetracosane 643-31-1 n-pentacosane 629-99-2 n-hexacosane 630-01-3 n-heptacosane 593-49-7 n-octacosane 630-02-4 n-nonacosane 630-03-5 n-triacontane 638-68-6 n-hentriacontane 630-04-6 n-dotriacontane 544-85-4 n-tritriacontane 630-05-7 n-tetratriacontane 14167-59-0 Table 2. Chemical names and alternatives, abbreviations, and Chemical Abstracts Service registry numbers.—Continued [Final list compiled according to the National Institute of Standards and Technology (NIST) Web site (http://webbook.nist.gov/chemistry/); NIST Standard Reference Database No. -

Global Journal of Science Frontier Research: B Chemistry
Online ISSN : 2249-4626 Print ISSN : 0975-5896 Chemistry and Medicinal Francolin and Turkey Theophylline and Caffeine Synthesis and Characterization VOLUME 13 ISSUE 3 VERSION 1.0 Global Journal of Science Frontier Research: B Chemistry Global Journal of Science Frontier Research: B C hemistry Vol ume 13 Issue 3 (Ver. 1.0) Open Association of Research Society © Global Journal of Science Global Journals Inc. Frontier Research .2013 . (A Delaware USA Incorporation with “Good Standing”; Reg. Number: 0423089) Sponsors: Open Association of Research Society All rights reserved. Open Scientific Standards This is a special issue published in version 1.0 of “Global Journal of Science Frontier Publisher’s Headquarters office Research.” By Global Journals Inc. All articles are open access articles distributed Global Journals Inc., Headquarters Corporate Office, under “Global Journal of Science Frontier Research” Cambridge Office Center, II Canal Park, Floor No. Reading License, which permits restricted use. 5th, Cambridge (Massachusetts), Pin: MA 02141 Entire contents are copyright by of “Global United States Journal of Science Frontier Research” unless USA Toll Free: +001-888-839-7392 otherwise noted on specific articles. USA Toll Free Fax: +001-888-839-7392 No part of this publication may be reproduced or transmitted in any form or by any means, Offset Typesetting electronic or mechanical, including photocopy, recording, or any information Open Association of Research Society, Marsh Road, storage and retrieval system, without written permission. Rainham, Essex, London RM13 8EU United Kingdom. The opinions and statements made in this book are those of the authors concerned. Ultraculture has not verified and neither confirms nor denies any of the foregoing and Packaging & Continental Dispatching no warranty or fitness is implied. -

Comparative Analysis of Volatile Compounds of Gamma-Irradiated Mutants of Rose (Rosa Hybrida)
plants Article Comparative Analysis of Volatile Compounds of Gamma-Irradiated Mutants of Rose (Rosa hybrida) Jaihyunk Ryu 1 , Jae Il Lyu 1 , Dong-Gun Kim 1, Jung-Min Kim 1, Yeong Deuk Jo 1 , Si-Yong Kang 2, Jin-Baek Kim 1 , Joon-Woo Ahn 1 and Sang Hoon Kim 1,* 1 Advanced Radiation Technology Institute, Korea Atomic Energy Research Institute, Jeollabuk-do 56212, Korea; [email protected] (J.R.); [email protected] (J.I.L.); [email protected] (D.-G.K.); [email protected] (J.-M.K.); [email protected] (Y.D.J.); [email protected] (J.-B.K.); [email protected] (J.-W.A.) 2 Department of Horticulture, College of Industrial Sciences, Kongju National University, Yesan 32439, Korea; [email protected] * Correspondence: [email protected]; Tel.: +82-63-570-3318 Received: 22 July 2020; Accepted: 14 September 2020; Published: 17 September 2020 Abstract: Roses are one of the most important floricultural crops, and their essential oils have long been used for cosmetics and aromatherapy. We investigated the volatile compound compositions of 12 flower-color mutant variants and their original cultivars. Twelve rose mutant genotypes were developed by treatment with 70 Gy of 60Co gamma irradiation of six commercial rose cultivars. Essential oils from the flowers of the 18 genotypes were analyzed by gas chromatography–mass spectrometry. Seventy-seven volatile compounds were detected, which were categorized into six classes: Aliphatic hydrocarbons, aliphatic alcohols, aliphatic ester, aromatic compounds, terpene alcohols, and others. Aliphatic (hydrocarbons, alcohols, and esters) compounds were abundant categories in all rose flowers. -

Exhaust Emission Profiles for EPA SPECIATE Database
Exhaust Emission Profiles for EPA SPECIATE Database: Energy Policy Act (EPAct) Low-Level Ethanol Fuel Blends and Tier 2 Light- Duty Vehicles Exhaust Emission Profiles for EPA SPECIATE Database: Energy Policy Act (EPAct) Low-Level Ethanol Fuel Blends and Tier 2 Light- Duty Vehicles Assessment and Standards Division Office of ransportationT and Air Quality U.S. Environmental Protection Agency EPA-420-R-09-002 June 2009 Table of Contents 1.0 Introduction....................................................................................................................... 1 1.1 Background..................................................................................................................... 1 1.2 Purpose of the Project ..................................................................................................... 1 2.0 Methods.............................................................................................................................. 2 2.1 Exhaust Emissions Data.................................................................................................. 2 2.2 Calculation of Composite Profiles.................................................................................. 3 2.3 Adjustment of E0 and E10 profiles with an In-Use Gasoline......................................... 3 2.4 Assignment of SPECIATE Identification Numbers ....................................................... 4 3.0 Results ............................................................................................................................... -

Oral Transfer of Chemical Cues, Growth Proteins and Hormones in Social Insects
Oral transfer of chemical cues, growth proteins and hormones in social insects LeBoeuf, A.C.1,2, Waridel, P.3, Brent, C.S.4, Gonçalves, A.N. 5,8, Menin, L. 6, Ortiz, D. 6, Riba- Grognuz, O. 2, Koto, A.7, Soares Z.G. 5,8, Privman, E. 9, Miska, E.A. 8,10,11, Benton, R.*1 and Keller, L.*2 * order decided on a coin-toss as these authors contributed equally. 1. Center for Integrative Genomics, University of Lausanne, CH-1015 Lausanne, Switzerland 2. Department of Ecology and Evolution, University of Lausanne, CH-1005 Lausanne, Switzerland 3. Protein Analysis Facility, University of Lausanne, 1015 Lausanne, Switzerland. 4. Arid Land Agricultural Research Center, USDA-ARS, 21881 N. Cardon Lane, Maricopa, Arizona, 85138, U.S.A. 5. Department of Biochemistry and Immunology, Instituto de Ciências Biológicas, Universidade Federal de Minas Gerais, Minas Gerais, CEP 30270-901, Brasil. 6. Institute of Chemical Sciences and Engineering, Ecole Polytechnique Fédérale de Lausanne, 1015- Lausanne, Switzerland. 7. The Department of Genetics, Graduate School of Pharmaceutical Sciences, The University of Tokyo. 8. Gurdon Institute, University of Cambridge, Tennis Court Road, Cambridge, CB2 1QN, United Kingdom 9. Department of Evolutionary and Environmental Biology, Institute of Evolution, University of Haifa, Haifa 3498838, Israel.Department of Genetics, University of Cambridge, Downing Street, Cambridge, CB2 3EH, United Kingdom 10. Wellcome Trust Sanger Institute, Wellcome Trust Genome Campus, Cambridge, CB10 1SA, United Kingdom Correspondence to: Adria C. LeBoeuf ([email protected]), Richard Benton ([email protected]) or Laurent Keller ([email protected]) 1 1 Abstract 2 3 Social insects frequently engage in oral fluid exchange – trophallaxis – between adults, and 4 between adults and larvae. -

UST and ASTM Petrochemical Standards
UST and ASTM Petrochemical Standards Your essential resource for Agilent ULTRA chemical standards Table of contents Introduction 3 Pennsylvania – GRO and PAH 22 About Agilent standards 3 Tennessee – GRO and DRO 22 Products 3 Texas – TNRCC Method 1005, 1006 23 Markets 3 Washington – Volatile Petroleum Hydrocarbons (VPH) method 24 Custom products 3 Washington – Extractable Petroleum Hydrocarbons Quality control laboratory 4 (EPH) method 25 Quality control validation levels 4 Washington and Oregon – Total Petroleum Hydrocarbons Triple certification 5 (NWTPH) methods 26 Level 2 reference material Certificate of Analysis 6 Maine – GRO and DRO 26 GHS compliance 7 Internal and surrogate standards for Underground Storage Tank (UST) testing 27 Underground Storage Tank (UST) Standards 8 EPA Method 1664A 28 Alaska – Method AK 101, AK 102, AK 103 10 EPA Method 418.1 28 Arizona – Method 8015AZ 11 Hydrocarbon fuel standards 29 California – PVOC and WIP 12 Weathered hydrocarbon fuel standards 30 Connecticut – ETPH method 12 EN 14105:2003 31 Florida – Method FL-PRO 13 Iowa – Methods OA-1, OA-2 14 ASTM Methods 32 Kansas – TPH method 15 ASTM Method D6584 32 Kansas modified 8015 (LRH) 15 ASTM Method D1387 33 Maine – Method 4.1.25, 4.2.17 16 ASTM Method D3710 34 Massachusetts – Volatile Petroleum Hydrocarbons ASTM Method D4815 34 (VPH) method 17 ASTM Method D5453 35/36 Massachusetts – Extractable Petroleum Hydrocarbons ASTM Methods D3120, D3246, D3961 36 (EPH) method 18 ASTM Method D4629 37 Shooters – Open and shoot spiking standards 19 ASTM Method D5762 38 Michigan – GRO and PNA 20 ASTM Method D4929 38 Mississippi – GRO, DRO, and PAH 20 ASTM Method D5808 38 New Jersey – OQA-QAM-025 21 New York – STARS compounds 22 Agilent Service and Support 39 2 UST and ASTM Petrochemical Standards Introduction About Agilent standards Agilent is a global leader in chromatography and spectroscopy, as well as an expert in chemical standards manufacturing. -

ABSTRACT RUDD, HAYDEN. Assessing the Vulnerability Of
ABSTRACT RUDD, HAYDEN. Assessing the Vulnerability of Coastal Plain Groundwater to Flood Water Intrusion using High Resolution Mass Spectrometry. (Under the direction of Dr. Elizabeth Guthrie Nichols). Communities in the North Carolina Coastal Plain (NCCP) depend on safe and reliable groundwater for private well use, agriculture, industry, and livelihoods. Although storm intensity and frequency are predicted to increase in coastal areas, the risk of surficial and confined aquifer contamination from extreme storms is not understood. In September 2018, Hurricane Florence caused extensive flooding across the NCCP for several weeks. The North Carolina Department of Environmental Quality (NCDEQ) Groundwater Management Branch had just completed sampling of some wells in their monitoring network when Hurricane Florence made landfall. NCDEQ returned to these wells, particularly those flooded by Hurricane Florence, for post-flood sampling. These groundwater samples were analyzed by NCDEQ for regulated semi-volatile organics with few to any detections of regulated organic contaminants. NCDEQ provided NC State the same sample extracts for analysis by high resolution mass spectrometry (HRMS). This research reports on the non-targeted and suspect-screening HRMS analyses of groundwater from nested monitoring wells. Some monitoring well sites experienced flooding during the study period, and some did not. The goal of this research was to advance our understanding of coastal aquifer susceptibility to flooding by producing the first comprehensive organic chemical profiles of coastal aquifers and by determining if aquifers have distinct organic chemical profiles that change after flooding events. This study used HRMS analyses to produce the first comprehensive organic chemical profiles of 11 aquifers in the coastal plain. -

November 30, 2017 Mr. William F. Durham Director WVDEP, Division
November 30, 2017 Mr. William F. Durham Director WVDEP, Division of Air Quality 601 – 57th Street SE Charleston, West Virginia 25304 Re: Tug Hill Operating, LLC, Permit Determination Application – Hendrickson Well Pad Dear Mr. Durham, Tug Hill Operating, LLC (Tug Hill) and SLR International Corporation (SLR) have prepared the attached Permit Determination Application for the Hendrickson Well Pad located in Marshall County, West Virginia. The site was purchased as a non-permitted pad below permitting thresholds and as a result, no DAQ ownership transfer forms are necessary and this should be the first determination submitted for the site. This determination reflects the addition of a Caterpillar G3508LE 4SLB compressor engine. The compressor engine is being proposed to lower the well’s operating pressure and boost pressure before entering the sales pipeline. Therefore, all site emissions have been evaluated and are attached for your review within this determination. If any additional information is needed, please feel free to contact me by telephone at (304) 545- 8563 or by e-mail at [email protected] Sincerely, SLR International Corporation Jesse Hanshaw, P.E. Principal Engineer SLR International Corporation, 8 Capitol Street, Suite 300, Charleston, WV 25301 681 205 8949 slrconsulting.com Tug Hill Operating, LLC Hendrickson Well Pad Proctor, West Virginia Permit Determination SLR Ref: 116.01631.00015 November 2017 Hendrickson Well Pad Permit Determination Prepared for: Tug Hill Operating, LLC 380 Southpointe Blvd., Suite 200 Canonsburg, PA 15317 This document has been prepared by SLR International Corporation. The material and data in this permit application were prepared under the supervision and direction of the undersigned. -

Two-Dimensional Gas Chromatography Characterization of Saw Palmetto Serenoa Repens Chemical Composition
United States Department of Agriculture Two-Dimensional Gas Chromatography Characterization of Saw Palmetto Serenoa repens Chemical Composition Roderquita K. Moore Doreen Mann Second dimension (s) dimension Second First dimension (s) Forest Forest Products Research Note September Service Laboratory FPL–RN–0387 2020 Abstract Contents Comprehensive two-dimensional gas chromatography Introduction ..........................................................................1 (GC×GC) is an important technique used to investigate Experimental ........................................................................1 potential medicinal mixtures. The pharmaceutical industry is a trillion-dollar industry, and 25% of this value comes Results and Discussion ........................................................2 from plant-derived chemicals. Saw Palmetto (Serenoa Conclusions ..........................................................................8 repens) berries are known for their medicinal properties. This medicinal alternative aids in treatment of health Acknowledgments ................................................................8 problems such as asthma, benign prostatic hypertrophy, References ..........................................................................10 colds, migraines, prostate cancer, and sore throats. Cancer is a disease where abnormal cells rapidly divide and destroy body tissue. In 2018, an estimated 1.7 million new cases of cancer were diagnosed in the United States and 609,640 people will die from the disease.