Plasma Assisted Decomposition of Methane and Propane and Cracking of Liquid Hexadecane
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Catalytic Pyrolysis of Plastic Wastes for the Production of Liquid Fuels for Engines
Electronic Supplementary Material (ESI) for RSC Advances. This journal is © The Royal Society of Chemistry 2019 Supporting information for: Catalytic pyrolysis of plastic wastes for the production of liquid fuels for engines Supattra Budsaereechaia, Andrew J. Huntb and Yuvarat Ngernyen*a aDepartment of Chemical Engineering, Faculty of Engineering, Khon Kaen University, Khon Kaen, 40002, Thailand. E-mail:[email protected] bMaterials Chemistry Research Center, Department of Chemistry and Center of Excellence for Innovation in Chemistry, Faculty of Science, Khon Kaen University, Khon Kaen, 40002, Thailand Fig. S1 The process for pelletization of catalyst PS PS+bentonite PP ) t e PP+bentonite s f f o % ( LDPE e c n a t t LDPE+bentonite s i m s n HDPE a r T HDPE+bentonite Gasohol 91 Diesel 4000 3500 3000 2500 2000 1500 1000 500 Wavenumber (cm-1) Fig. S2 FTIR spectra of oil from pyrolysis of plastic waste type. Table S1 Compounds in oils (%Area) from the pyrolysis of plastic wastes as detected by GCMS analysis PS PP LDPE HDPE Gasohol 91 Diesel Compound NC C Compound NC C Compound NC C Compound NC C 1- 0 0.15 Pentane 1.13 1.29 n-Hexane 0.71 0.73 n-Hexane 0.65 0.64 Butane, 2- Octane : 0.32 Tetradecene methyl- : 2.60 Toluene 7.93 7.56 Cyclohexane 2.28 2.51 1-Hexene 1.05 1.10 1-Hexene 1.15 1.16 Pentane : 1.95 Nonane : 0.83 Ethylbenzen 15.07 11.29 Heptane, 4- 1.81 1.68 Heptane 1.26 1.35 Heptane 1.22 1.23 Butane, 2,2- Decane : 1.34 e methyl- dimethyl- : 0.47 1-Tridecene 0 0.14 2,2-Dimethyl- 0.63 0 1-Heptene 1.37 1.46 1-Heptene 1.32 1.35 Pentane, -

Alteration of Volatile Chemical Composition in Tobacco Plants Due to Green Peach Aphid (Myzus Persicae Sulzer) (Hemiptera: Aphididae) Feeding
Song et al.: Green peach aphid (Myzus persicae Sulzer) (Hemiptera: Aphididae) feeding alters volatile composition in tobacco plants - 159 - ALTERATION OF VOLATILE CHEMICAL COMPOSITION IN TOBACCO PLANTS DUE TO GREEN PEACH APHID (MYZUS PERSICAE SULZER) (HEMIPTERA: APHIDIDAE) FEEDING SONG, Y. Z.1,2,3 – GUO, Y. Q.1,2 – CAI, P. M.2,3 – CHEN, W. B.1,2 – LIU, C. M.1,2* 1Biological Control Research Institute, College of Plant Protection, Fujian Agriculture and Forestry University, Fuzhou 350002, China e-mail: [email protected] (Song, Y. Z.); [email protected] (Guo, Y. Q.); [email protected] (Chen, W. B.) 2State Key Laboratory of Ecological Pest Control for Fujian and Taiwan Crops, Fuzhou 350002, China e-mail: [email protected] (Cai, P. M.) 3Department of Horticulture, College of Tea and Food Science, Wuyi University, Wuyishan 354300, China *Corresponding author e-mail: [email protected]; phone: +86-0591-8378-9420; fax: +86-0591-8378-9421 (Received 10th Jul 2020; accepted 6th Oct 2020) Abstract. In response to insect pest herbivory, plants can generate volatile components that may serve multiple roles as communication signals and defence agents in a multitrophic context. In the present study, the volatile profiles of tobacco plants Nicotiana tabacum L., with and without infestation by sap-sucking aphids Myzus persicae Sulzer, were measured by gas chromatography-mass spectrometry (GC-MS). The results revealed that a total of 10 compounds were identified from healthy tobacco plants, and a total of 14 and 16 compounds were isolated from aphid-infested tobacco plants at 24 and 48 hours after infestation, respectively. -

N-Hexadecane Fuel for a Phosphoric Acid Direct Hydrocarbon Fuel Cell
Hindawi Publishing Corporation Journal of Fuels Volume 2015, Article ID 748679, 9 pages http://dx.doi.org/10.1155/2015/748679 Research Article n-Hexadecane Fuel for a Phosphoric Acid Direct Hydrocarbon Fuel Cell Yuanchen Zhu,1,2 Travis Robinson,1 Amani Al-Othman,2,3 André Y. Tremblay,1 and Marten Ternan4 1 Chemical and Biological Engineering, University of Ottawa, 161 Louis Pasteur, Ottawa, ON, Canada K1N 6N5 2Catalysis Centre for Research and Innovation, University of Ottawa, 30 Marie Curie, Ottawa, ON, Canada K1N 6N5 3Chemical Engineering, American University of Sharjah, Sharjah, UAE 4EnPross Incorporated, 147 Banning Road, Ottawa, ON, Canada K2L 1C5 Correspondence should be addressed to Marten Ternan; [email protected] Received 8 January 2015; Accepted 17 March 2015 Academic Editor: Michele Gambino Copyright © 2015 Yuanchen Zhu et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. The objective of this work was to examine fuel cells as a possible alternative to the diesel fuel engines currently used inrailway locomotives, thereby decreasing air emissions from the railway transportation sector. We have investigated the performance of a phosphoric acid fuel cell (PAFC) reactor, with n-hexadecane, C16H34 (a model compound for diesel fuel, cetane number = 100). This is the first extensive study reported in the literature in which n-hexadecane is used directly as the fuel. Measurements were made to obtain both polarization curves and time-on-stream results. Because deactivation was observed hydrogen polarization curves were measured before and after n-hexadecane experiments, to determine the extent of deactivation of the membrane electrode assembly (MEA). -

Synthetic Turf Scientific Advisory Panel Meeting Materials
California Environmental Protection Agency Office of Environmental Health Hazard Assessment Synthetic Turf Study Synthetic Turf Scientific Advisory Panel Meeting May 31, 2019 MEETING MATERIALS THIS PAGE LEFT BLANK INTENTIONALLY Office of Environmental Health Hazard Assessment California Environmental Protection Agency Agenda Synthetic Turf Scientific Advisory Panel Meeting May 31, 2019, 9:30 a.m. – 4:00 p.m. 1001 I Street, CalEPA Headquarters Building, Sacramento Byron Sher Auditorium The agenda for this meeting is given below. The order of items on the agenda is provided for general reference only. The order in which items are taken up by the Panel is subject to change. 1. Welcome and Opening Remarks 2. Synthetic Turf and Playground Studies Overview 4. Synthetic Turf Field Exposure Model Exposure Equations Exposure Parameters 3. Non-Targeted Chemical Analysis Volatile Organics on Synthetic Turf Fields Non-Polar Organics Constituents in Crumb Rubber Polar Organic Constituents in Crumb Rubber 5. Public Comments: For members of the public attending in-person: Comments will be limited to three minutes per commenter. For members of the public attending via the internet: Comments may be sent via email to [email protected]. Email comments will be read aloud, up to three minutes each, by staff of OEHHA during the public comment period, as time allows. 6. Further Panel Discussion and Closing Remarks 7. Wrap Up and Adjournment Agenda Synthetic Turf Advisory Panel Meeting May 31, 2019 THIS PAGE LEFT BLANK INTENTIONALLY Office of Environmental Health Hazard Assessment California Environmental Protection Agency DRAFT for Discussion at May 2019 SAP Meeting. Table of Contents Synthetic Turf and Playground Studies Overview May 2019 Update ..... -

The Enthalpy of Formation of Organic Compounds with “Chemical Accuracy”
chemengineering Article Group Contribution Revisited: The Enthalpy of Formation of Organic Compounds with “Chemical Accuracy” Robert J. Meier Pro-Deo Consultant, 52525 Heinsberg, North-Rhine Westphalia, Germany; [email protected] Abstract: Group contribution (GC) methods to predict thermochemical properties are of eminent importance to process design. Compared to previous works, we present an improved group contri- bution parametrization for the heat of formation of organic molecules exhibiting chemical accuracy, i.e., a maximum 1 kcal/mol (4.2 kJ/mol) difference between the experiment and model, while, at the same time, minimizing the number of parameters. The latter is extremely important as too many parameters lead to overfitting and, therewith, to more or less serious incorrect predictions for molecules that were not within the data set used for parametrization. Moreover, it was found to be important to explicitly account for common chemical knowledge, e.g., geminal effects or ring strain. The group-related parameters were determined step-wise: first, alkanes only, and then only one additional group in the next class of molecules. This ensures unique and optimal parameter values for each chemical group. All data will be made available, enabling other researchers to extend the set to other classes of molecules. Keywords: enthalpy of formation; thermodynamics; molecular modeling; group contribution method; quantum mechanical method; chemical accuracy; process design Citation: Meier, R.J. Group Contribution Revisited: The Enthalpy of Formation of Organic Compounds with “Chemical Accuracy”. 1. Introduction ChemEngineering 2021, 5, 24. To understand chemical reactivity and/or chemical equilibria, knowledge of thermo- o https://doi.org/10.3390/ dynamic properties such as gas-phase standard enthalpy of formation DfH gas is a necessity. -

Vapor Pressures and Vaporization Enthalpies of the N-Alkanes from C31 to C38 at T ) 298.15 K by Correlation Gas Chromatography
BATCH: je3a29 USER: ckt69 DIV: @xyv04/data1/CLS_pj/GRP_je/JOB_i03/DIV_je030236t DATE: March 15, 2004 1 Vapor Pressures and Vaporization Enthalpies of the n-Alkanes from 2 C31 to C38 at T ) 298.15 K by Correlation Gas Chromatography 3 James S. Chickos* and William Hanshaw 4 Department of Chemistry and Biochemistry, University of MissourisSt. Louis, St. Louis, Missouri 63121, USA 5 6 The temperature dependence of gas-chromatographic retention times for henitriacontane to octatriacontane 7 is reported. These data are used in combination with other literature values to evaluate the vaporization 8 enthalpies and vapor pressures of these n-alkanes from T ) 298.15 to 575 K. The vapor pressure and 9 vaporization enthalpy results obtained are compared with existing literature data where possible and 10 found to be internally consistent. The vaporization enthalpies and vapor pressures obtained for the 11 n-alkanes are also tested directly and through the use of thermochemical cycles using literature values 12 for several polyaromatic hydrocarbons. Sublimation enthalpies are also calculated for hentriacontane to 13 octatriacontane by combining vaporization enthalpies with temperature adjusted fusion enthalpies. 14 15 Introduction vaporization enthalpies of C21 to C30 to include C31 to C38. 54 The vapor pressures and vaporization enthalpies of these 55 16 Polycyclic aromatic hydrocarbons (PAHs) are important larger n-alkanes have been evaluated using the technique 56 17 environmental contaminants, many of which exhibit very of correlation gas chromatography. This technique relies 57 18 low vapor pressures. In the gas phase, they exist mainly entirely on the use of standards in assessment. Standards 58 19 sorbed to aerosol particles. -

Measurements of Higher Alkanes Using NO Chemical Ionization in PTR-Tof-MS
Atmos. Chem. Phys., 20, 14123–14138, 2020 https://doi.org/10.5194/acp-20-14123-2020 © Author(s) 2020. This work is distributed under the Creative Commons Attribution 4.0 License. Measurements of higher alkanes using NOC chemical ionization in PTR-ToF-MS: important contributions of higher alkanes to secondary organic aerosols in China Chaomin Wang1,2, Bin Yuan1,2, Caihong Wu1,2, Sihang Wang1,2, Jipeng Qi1,2, Baolin Wang3, Zelong Wang1,2, Weiwei Hu4, Wei Chen4, Chenshuo Ye5, Wenjie Wang5, Yele Sun6, Chen Wang3, Shan Huang1,2, Wei Song4, Xinming Wang4, Suxia Yang1,2, Shenyang Zhang1,2, Wanyun Xu7, Nan Ma1,2, Zhanyi Zhang1,2, Bin Jiang1,2, Hang Su8, Yafang Cheng8, Xuemei Wang1,2, and Min Shao1,2 1Institute for Environmental and Climate Research, Jinan University, 511443 Guangzhou, China 2Guangdong-Hongkong-Macau Joint Laboratory of Collaborative Innovation for Environmental Quality, 511443 Guangzhou, China 3School of Environmental Science and Engineering, Qilu University of Technology (Shandong Academy of Sciences), 250353 Jinan, China 4State Key Laboratory of Organic Geochemistry and Guangdong Key Laboratory of Environmental Protection and Resources Utilization, Guangzhou Institute of Geochemistry, Chinese Academy of Sciences, 510640 Guangzhou, China 5State Joint Key Laboratory of Environmental Simulation and Pollution Control, College of Environmental Sciences and Engineering, Peking University, 100871 Beijing, China 6State Key Laboratory of Atmospheric Boundary Physics and Atmospheric Chemistry, Institute of Atmospheric Physics, Chinese -
Supporting Information for Modeling the Formation and Composition Of
Supporting Information for Modeling the Formation and Composition of Secondary Organic Aerosol from Diesel Exhaust Using Parameterized and Semi-explicit Chemistry and Thermodynamic Models Sailaja Eluri1, Christopher D. Cappa2, Beth Friedman3, Delphine K. Farmer3, and Shantanu H. Jathar1 1 Department of Mechanical Engineering, Colorado State University, Fort Collins, CO, USA, 80523 2 Department of Civil and Environmental Engineering, University of California Davis, Davis, CA, USA, 95616 3 Department of Chemistry, Colorado State University, Fort Collins, CO, USA, 80523 Correspondence to: Shantanu H. Jathar ([email protected]) Table S1: Mass speciation and kOH for VOC emissions profile #3161 3 -1 - Species Name kOH (cm molecules s Mass Percent (%) 1) (1-methylpropyl) benzene 8.50×10'() 0.023 (2-methylpropyl) benzene 8.71×10'() 0.060 1,2,3-trimethylbenzene 3.27×10'(( 0.056 1,2,4-trimethylbenzene 3.25×10'(( 0.246 1,2-diethylbenzene 8.11×10'() 0.042 1,2-propadiene 9.82×10'() 0.218 1,3,5-trimethylbenzene 5.67×10'(( 0.088 1,3-butadiene 6.66×10'(( 0.088 1-butene 3.14×10'(( 0.311 1-methyl-2-ethylbenzene 7.44×10'() 0.065 1-methyl-3-ethylbenzene 1.39×10'(( 0.116 1-pentene 3.14×10'(( 0.148 2,2,4-trimethylpentane 3.34×10'() 0.139 2,2-dimethylbutane 2.23×10'() 0.028 2,3,4-trimethylpentane 6.60×10'() 0.009 2,3-dimethyl-1-butene 5.38×10'(( 0.014 2,3-dimethylhexane 8.55×10'() 0.005 2,3-dimethylpentane 7.14×10'() 0.032 2,4-dimethylhexane 8.55×10'() 0.019 2,4-dimethylpentane 4.77×10'() 0.009 2-methylheptane 8.28×10'() 0.028 2-methylhexane 6.86×10'() -

Safety Data Sheet
SAFETY DATA SHEET Flammable Liquid Mixture: Docosane / Dodecane / Eicosane / Hexacosane / Hexadecane / Hexane / N-Decane / N-Heptane / N-Nonane / N-Octadecane / N- Octane / Octacosane / Tetracosane / Tetradecane Section 1. Identification GHS product identifier : Flammable Liquid Mixture: Docosane / Dodecane / Eicosane / Hexacosane / Hexadecane / Hexane / N-Decane / N-Heptane / N-Nonane / N-Octadecane / N-Octane / Octacosane / Tetracosane / Tetradecane Other means of : Not available. identification Product use : Synthetic/Analytical chemistry. SDS # : 019735 Supplier's details : Airgas USA, LLC and its affiliates 259 North Radnor-Chester Road Suite 100 Radnor, PA 19087-5283 1-610-687-5253 24-hour telephone : 1-866-734-3438 Section 2. Hazards identification OSHA/HCS status : This material is considered hazardous by the OSHA Hazard Communication Standard (29 CFR 1910.1200). Classification of the : FLAMMABLE LIQUIDS - Category 1 substance or mixture SKIN CORROSION/IRRITATION - Category 2 SERIOUS EYE DAMAGE/ EYE IRRITATION - Category 2A TOXIC TO REPRODUCTION (Fertility) - Category 2 TOXIC TO REPRODUCTION (Unborn child) - Category 2 SPECIFIC TARGET ORGAN TOXICITY (SINGLE EXPOSURE) (Respiratory tract irritation) - Category 3 SPECIFIC TARGET ORGAN TOXICITY (SINGLE EXPOSURE) (Narcotic effects) - Category 3 SPECIFIC TARGET ORGAN TOXICITY (REPEATED EXPOSURE) - Category 2 AQUATIC HAZARD (ACUTE) - Category 2 AQUATIC HAZARD (LONG-TERM) - Category 1 GHS label elements Hazard pictograms : Signal word : Danger Hazard statements : Extremely flammable liquid and vapor. May form explosive mixtures in Air. Causes serious eye irritation. Causes skin irritation. May cause respiratory irritation. Suspected of damaging fertility or the unborn child. May cause drowsiness and dizziness. May cause damage to organs through prolonged or repeated exposure. Very toxic to aquatic life with long lasting effects. Precautionary statements General : Read label before use. -

Vapor Pressures and Vaporization Enthalpies of the N-Alkanes from 2 C21 to C30 at T ) 298.15 K by Correlation Gas Chromatography
BATCH: je1a04 USER: jeh69 DIV: @xyv04/data1/CLS_pj/GRP_je/JOB_i01/DIV_je0301747 DATE: October 17, 2003 1 Vapor Pressures and Vaporization Enthalpies of the n-Alkanes from 2 C21 to C30 at T ) 298.15 K by Correlation Gas Chromatography 3 James S. Chickos* and William Hanshaw 4 Department of Chemistry and Biochemistry, University of MissourisSt. Louis, St. Louis, Missouri 63121 5 6 The temperature dependence of gas chromatographic retention times for n-heptadecane to n-triacontane 7 is reported. These data are used to evaluate the vaporization enthalpies of these compounds at T ) 298.15 8 K, and a protocol is described that provides vapor pressures of these n-alkanes from T ) 298.15 to 575 9 K. The vapor pressure and vaporization enthalpy results obtained are compared with existing literature 10 data where possible and found to be internally consistent. Sublimation enthalpies for n-C17 to n-C30 are 11 calculated by combining vaporization enthalpies with fusion enthalpies and are compared when possible 12 to direct measurements. 13 14 Introduction 15 The n-alkanes serve as excellent standards for the 16 measurement of vaporization enthalpies of hydrocarbons.1,2 17 Recently, the vaporization enthalpies of the n-alkanes 18 reported in the literature were examined and experimental 19 values were selected on the basis of how well their 20 vaporization enthalpies correlated with their enthalpies of 21 transfer from solution to the gas phase as measured by gas 22 chromatography.3 A plot of the vaporization enthalpies of 23 the n-alkanes as a function of the number of carbon atoms 24 is given in Figure 1. -
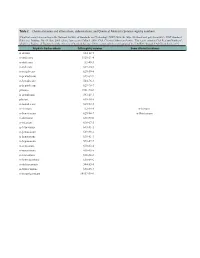
Table 2. Chemical Names and Alternatives, Abbreviations, and Chemical Abstracts Service Registry Numbers
Table 2. Chemical names and alternatives, abbreviations, and Chemical Abstracts Service registry numbers. [Final list compiled according to the National Institute of Standards and Technology (NIST) Web site (http://webbook.nist.gov/chemistry/); NIST Standard Reference Database No. 69, June 2005 release, last accessed May 9, 2008. CAS, Chemical Abstracts Service. This report contains CAS Registry Numbers®, which is a Registered Trademark of the American Chemical Society. CAS recommends the verification of the CASRNs through CAS Client ServicesSM] Aliphatic hydrocarbons CAS registry number Some alternative names n-decane 124-18-5 n-undecane 1120-21-4 n-dodecane 112-40-3 n-tridecane 629-50-5 n-tetradecane 629-59-4 n-pentadecane 629-62-9 n-hexadecane 544-76-3 n-heptadecane 629-78-7 pristane 1921-70-6 n-octadecane 593-45-3 phytane 638-36-8 n-nonadecane 629-92-5 n-eicosane 112-95-8 n-Icosane n-heneicosane 629-94-7 n-Henicosane n-docosane 629-97-0 n-tricosane 638-67-5 n-tetracosane 643-31-1 n-pentacosane 629-99-2 n-hexacosane 630-01-3 n-heptacosane 593-49-7 n-octacosane 630-02-4 n-nonacosane 630-03-5 n-triacontane 638-68-6 n-hentriacontane 630-04-6 n-dotriacontane 544-85-4 n-tritriacontane 630-05-7 n-tetratriacontane 14167-59-0 Table 2. Chemical names and alternatives, abbreviations, and Chemical Abstracts Service registry numbers.—Continued [Final list compiled according to the National Institute of Standards and Technology (NIST) Web site (http://webbook.nist.gov/chemistry/); NIST Standard Reference Database No. -

The Strength in Numbers: Comprehensive Characterization of House Dust Using Complementary Mass Spectrometric Techniques
Analytical and Bioanalytical Chemistry https://doi.org/10.1007/s00216-019-01615-6 PAPER IN FOREFRONT The strength in numbers: comprehensive characterization of house dust using complementary mass spectrometric techniques Pawel Rostkowski1 & Peter Haglund2 & Reza Aalizadeh3 & Nikiforos Alygizakis 3,4 & Nikolaos Thomaidis 3 & Joaquin Beltran Arandes5 & Pernilla Bohlin Nizzetto1 & Petra Booij 6 & Hélène Budzinski7 & Pamela Brunswick8 & Adrian Covaci9 & Christine Gallampois2 & Sylvia Grosse10 & Ralph Hindle11 & Ildiko Ipolyi4 & Karl Jobst12 & Sarit L. Kaserzon13 & Pim Leonards14 & Francois Lestremau15 & Thomas Letzel10 & Jörgen Magnér16,17 & Hidenori Matsukami18 & Christoph Moschet19 & Peter Oswald 4 & Merle Plassmann20 & Jaroslav Slobodnik4 & Chun Yang21 Received: 6 November 2018 /Revised: 20 December 2018 /Accepted: 15 January 2019 # The Author(s) 2019 Abstract Untargeted analysis of a composite house dust sample has been performed as part of a collaborative effort to evaluate the progress in the field of suspect and nontarget screening and build an extensive database of organic indoor environment contaminants. Twenty- one participants reported results that were curated by the organizers of the collaborative trial. In total, nearly 2350 compounds were identified (18%) or tentatively identified (25% at confidence level 2 and 58% at confidence level 3), making the collaborative trial a success. However, a relatively small share (37%) of all compounds were reported by more than one participant, which shows that there is plenty of room for improvement in the field of suspect and nontarget screening. An even a smaller share (5%) of the total number of compounds were detected using both liquid chromatography–mass spectrometry (LC-MS) and gas chromatography– mass spectrometry (GC-MS). Thus, the two MS techniques are highly complementary.