Rutherfordium (Element 104)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Development of the Periodic Table and Its Consequences Citation: J
Firenze University Press www.fupress.com/substantia The Development of the Periodic Table and its Consequences Citation: J. Emsley (2019) The Devel- opment of the Periodic Table and its Consequences. Substantia 3(2) Suppl. 5: 15-27. doi: 10.13128/Substantia-297 John Emsley Copyright: © 2019 J. Emsley. This is Alameda Lodge, 23a Alameda Road, Ampthill, MK45 2LA, UK an open access, peer-reviewed article E-mail: [email protected] published by Firenze University Press (http://www.fupress.com/substantia) and distributed under the terms of the Abstract. Chemistry is fortunate among the sciences in having an icon that is instant- Creative Commons Attribution License, ly recognisable around the world: the periodic table. The United Nations has deemed which permits unrestricted use, distri- 2019 to be the International Year of the Periodic Table, in commemoration of the 150th bution, and reproduction in any medi- anniversary of the first paper in which it appeared. That had been written by a Russian um, provided the original author and chemist, Dmitri Mendeleev, and was published in May 1869. Since then, there have source are credited. been many versions of the table, but one format has come to be the most widely used Data Availability Statement: All rel- and is to be seen everywhere. The route to this preferred form of the table makes an evant data are within the paper and its interesting story. Supporting Information files. Keywords. Periodic table, Mendeleev, Newlands, Deming, Seaborg. Competing Interests: The Author(s) declare(s) no conflict of interest. INTRODUCTION There are hundreds of periodic tables but the one that is widely repro- duced has the approval of the International Union of Pure and Applied Chemistry (IUPAC) and is shown in Fig.1. -

Tables of Parameters Tables of Parameters for Extended Hückel Calculations Calculations
TABLES OF PARAMETERS FOR EXTENDED HÜCKEL CALCULATIONS Santiago Alvarez Universitat de Barcelona 1999 - 1 - PART 1. STANDARD Hii's and SLATER EXPONENTS AND COEFFICIENTS. H He Li Be B C N O F Ne Na Mg Al Si P S Cl Ar K Ca Sc Ti V Cr Mn Fe Co Ni Cu Zn Ga Ge As Se Br Kr Rb Sr Y Zr Nb Mo Tc Ru Rh Pd Ag Cd In Sn Sb Te I Xe Cs Ba Hf Ta W Re Os Ir Pt Au Hg Tl Pb Bi Po At Rn Fr Ra Rf Db Sg Bh Hs Mt La Ce Pr Nd Pm Sm Eu Gd Tb Dy Ho Er Tm Yb Lu Ac Th Pa U Np Pu Am Cm Bk Cf Es Fm Md No Lr Parameters for all elements except the shaded ones are included in these Tables. - 2 - General Observations References are to the original paper which first reported EH calculations with the given Hii for a related set of molecules or solids. For some elements, when no EH calculations have been reported, the experimental ionization potentials are given and the references correspond to those experimental values. Although these tables are intended to be of some use in locating atomic parameters, reference to the original work is encouraged. Ionization potentials ( Hii) a) All energies are in eV. Hii's for most transition metals are charge-iterated for model compounds like metal carbonyls or typical organometallic molecules with Cp, CH3, CO, etc. Exceptions, including bulk metals are conveniently indicated. For cases in which charge-iterated Hii's are not available, the standard sources are the Hinze and Jaffé's tables of ionization potentials.1 b) When alternative sets of Hii's were available for the same element, the one which fits better with the trend along a group or a period of the periodic table has been chosen. -
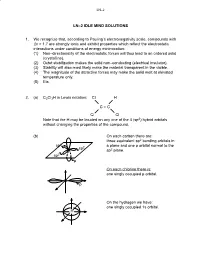
LN–2 IDLE MIND SOLUTIONS 1. We Recognize That, According To
LN–2 LN–2 IDLE MIND SOLUTIONS 1. We recognize that, according to Pauling’s electronegativity scale, compounds with Δx > 1.7 are strongly ionic and exhibit properties which reflect the electrostatic interactions under conditions of energy minimization: (1) Non–directionality of the electrostatic forces will thus lead to an ordered solid (crystalline). (2) Octet stabilization makes the solid non–conducting (electrical insulator). (3) Stability will also most likely make the material transparent in the visible. (4) The magnitude of the attractive forces may make the solid melt at elevated temperature only. (5) Etc. 2. (a) C2Cl3H in Lewis notation: Cl H C = C Cl Cl Note that the H may be located on any one of the 4 (sp2) hybrid orbitals without changing the properties of the compound. (b) On each carbon there are: three equivalent sp2 bonding orbitals in sp2 a plane and one p orbital normal to the sp2 sp2 plane. sp2 p On each chlorine there is: one singly occupied p orbital. Cl p On the hydrogen we have: one singly occupied 1s orbital. H LN–2 σ 2. (c) 2 sp2–sp2 orbitals → 1σ C–C π 2 p–p orbitals → 1π C–C 6 bonds 3 sp2–p orbitals → 3σ C–Cl 1 sp2–s orbital → 1σ C–H 3. Silicon (Si) is a group 4 element like C (carbon) and wil also exhibit sp3 hybridization: 4 sp3 orbitals at B.A. = 109o. Unlike carbon, silicon cannot form double or triple bonds (by lateral p orbital overlap) because the atom is too large to permit lateral overlap! (a) Si + 2Cl2 → SiCl4 Si is in the center of a tetrahedron with the chlorine atoms as the four corners. -

Week 1 Slides
Adventures in Chemistry Instructor: Abi Oyeyemi Week 1 States of matter Boiling point, Melting point Atoms Types of Chemistry Hazards PPE Atoms consist of: Neutrons Protons Electrons 0 + - In In shells of atoms Nucleus States of Matter Solid, liquid, gas Week 2 Flame tests Orbitals Periodic table Octet Groups and Periods Experiment: Making a non-Newtonian Fluid Periodic Table Groups Each group tells how many electrons are in the outer shell. In chemistry, a group (also known as a family) is a column of elements in the periodic table of the chemical elements. The elements in each group have the same number of electrons in the outer orbital. Those outer electrons are also called valence electrons Periodic table 118 elements total 8 groups Group 1- Alkali metals Group 2- Alkali earth metals Group 7 –Halogens Group 8-Noble gases Elements structure Includes abbreviated name of element Atomic number-number of protons Always same number of protons and neutrons Mass number- average no. of neutrons and protons Amu: atomic mass units Isotopes Periodic table: Periods A period in the periodic table is a row of chemical elements. All elements in a row have the same number of electron shells/orbitals Week 3 More on the periodic table Electronic configuration Isotopes Chemical vs Physical changes Compounds vs Mixtures Ionic and Covalent bonds Demonstration Lava Lamp Activity: Making models Why do elements react? Octet, want a full outer shell How? By taking or giving electrons (ionic bonding) Or by sharing electrons (covalent bonding) Compounds- A compound is a substance formed when two or more chemical elements are chemically bonded together. -

Periodic Table 1 Periodic Table
Periodic table 1 Periodic table This article is about the table used in chemistry. For other uses, see Periodic table (disambiguation). The periodic table is a tabular arrangement of the chemical elements, organized on the basis of their atomic numbers (numbers of protons in the nucleus), electron configurations , and recurring chemical properties. Elements are presented in order of increasing atomic number, which is typically listed with the chemical symbol in each box. The standard form of the table consists of a grid of elements laid out in 18 columns and 7 Standard 18-column form of the periodic table. For the color legend, see section Layout, rows, with a double row of elements under the larger table. below that. The table can also be deconstructed into four rectangular blocks: the s-block to the left, the p-block to the right, the d-block in the middle, and the f-block below that. The rows of the table are called periods; the columns are called groups, with some of these having names such as halogens or noble gases. Since, by definition, a periodic table incorporates recurring trends, any such table can be used to derive relationships between the properties of the elements and predict the properties of new, yet to be discovered or synthesized, elements. As a result, a periodic table—whether in the standard form or some other variant—provides a useful framework for analyzing chemical behavior, and such tables are widely used in chemistry and other sciences. Although precursors exist, Dmitri Mendeleev is generally credited with the publication, in 1869, of the first widely recognized periodic table. -
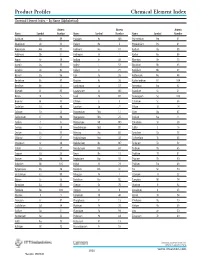
Product Profiles Chemical Element Index
Product Profiles Chemical Element Index Chemical Element Index – By Name (Alphabetical) Atomic Atomic Atomic Name Symbol Number Name Symbol Number Name Symbol Number Actinium Ac 89 Hassium Hs 108 Promethium Pm 61 Aluminum Al 13 Helium He 2 Protactinium Pa 91 Americium Am 95 Holmium Ho 67 Radium Ra 88 Antimony Sb 51 Hydrogen H 1 Radon Rn 86 Argon Ar 18 Indium In 49 Rhenium Re 75 Arsenic As 33 Iodine I 53 Rhodium Rh 45 Astatine At 85 Iridium Ir 77 Rubidium Rb 37 Barium Ba 56 Iron Fe 26 Ruthenium Ru 44 Berkelium Bk 97 Krypton Kr 36 Rutherfordium Rf 104 Beryllium Be 4 Lanthanum La 57 Samarium Sm 62 Bismuth Bi 83 Lawrencium Lr 103 Scandium Sc 21 Boron B 5 Lead Pb 82 Seaborgium Sg 106 Bromine Br 35 Lithium Li 3 Selenium Se 34 Cadmium Cd 48 Lutetium Lu 71 Silicon Si 14 Calcium Ca 20 Magnesium Mg 12 Silver Ag 47 Californium Cf 98 Manganese Mn 25 Sodium Na 11 Carbon C 6 Meitnerium Mt 109 Strontium Sr 38 Cerium Ce 58 Mendelevium Md 101 Sulfur S 16 Cesium Cs 55 Mercury Hg 80 Tantalum Ta 73 Chlorine Cl 17 Molybdenum Mo 42 Technetium Tc 43 Chromium Cr 24 Neilsborium Ns 107 Tellurium Te 52 Cobalt Co 27 Neodymium Nd 60 Terbium Tb 65 Copper Cu 29 Neon Ne 10 Thallium Tl 81 Curium Cm 96 Neptunium Np 93 Thorium Th 90 Dubnium Db 105 Nickel Ni 28 Thulium Tm 69 Dysprosium Dy 66 Niobium Nb 41 Tin Sn 50 Einsteinium Es 99 Nitrogen N 7 Titanium Ti 22 Erbium Er 68 Nobelium No 102 Tungsten W 74 Europium Eu 63 Osmian Os 76 Uranium U 92 Fermium Fm 100 Oxygen O 8 Vanadium V 23 Fluorine F 9 Palladium Pd 46 Xenon Xi 54 Francium Fr 87 Phosphorus P 15 Ytterbium Yb 70 Gadolinium -

Chemistry of Superheavy Elements Matthias Schädel*
Reviews M. Schädel DOI: 10.1002/anie.200461072 Superheavy Elements Chemistry of Superheavy Elements Matthias Schädel* Keywords: Dedicated to Professor Günter Herrmann atom-at-a-time chemistry · periodic on the occasion of his 80th birthday table · relativistic effects · superheavy elements · transactinides Angewandte Chemie 368 www.angewandte.org 2006 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Angew. Chem. Int. Ed. 2006, 45, 368 – 401 Angewandte Superheavy Elements Chemie The number of chemical elements has increased considerably in the From the Contents last few decades.Most excitingly, these heaviest, man-made elements at the far-end of the Periodic Table are located in the area of the long- 1. Introduction and Historical Remarks 369 awaited superheavy elements.While physical techniques currently play a leading role in these discoveries, the chemistry of superheavy 2. Nuclear Aspects 372 elements is now beginning to be developed.Advanced and very sensitive techniques allow the chemical properties of these elusive 3. Atom-at-a-Time Chemistry 374 elements to be probed.Often, less than ten short-lived atoms, chemi- 4. Objectives for Superheavy cally separated one-atom-at-a-time, provide crucial information on Element Chemistry 375 basic chemical properties.These results place the architecture of the far-end of the Periodic Table on the test bench and probe the 5. Experimental Techniques 376 increasingly strong relativistic effects that influence the chemical 6. Chemical Properties 380 properties there.This review is focused mainly on the experimental work on superheavy element chemistry.It contains a short contribu- 7. Summary and Perspectives 394 tion on relativistic theory, and some important historical and nuclear aspects. -

WO 2017/161171 A2 21 September 2017 (21.09.2017) P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2017/161171 A2 21 September 2017 (21.09.2017) P O P C T (51) International Patent Classification: (81) Designated States (unless otherwise indicated, for every B01J 23/10 (2006.01) B82Y 30/00 (201 1.01) kind of national protection available): AE, AG, AL, AM, B01J 35/02 (2006.01) C07C 2/84 (2006.01) AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DJ, DK, DM, (21) Number: International Application DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, PCT/US20 17/022787 HN, HR, HU, ID, IL, IN, IR, IS, JP, KE, KG, KH, KN, (22) International Filing Date: KP, KR, KW, KZ, LA, LC, LK, LR, LS, LU, LY, MA, 16 March 2017 (16.03.2017) MD, ME, MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, OM, PA, PE, PG, PH, PL, PT, QA, RO, RS, (25) Filing Language: English RU, RW, SA, SC, SD, SE, SG, SK, SL, SM, ST, SV, SY, (26) Publication Language: English TH, TJ, TM, TN, TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. (30) Priority Data: 62/309,284 16 March 2016 (16.03.2016) US (84) Designated States (unless otherwise indicated, for every kind of regional protection available): ARIPO (BW, GH, (71) Applicant: SILURIA TECHNOLOGIES, INC. GM, KE, LR, LS, MW, MZ, NA, RW, SD, SL, ST, SZ, [US/US]; 409 Illinois Street, Suite 5032, San Francisco, TZ, UG, ZM, ZW), Eurasian (AM, AZ, BY, KG, KZ, RU, California 94158 (US). -

Syntheses, Structures, Optical Properties, and Theoretical Calculations of Cs2bi2zns5,Cs2bi2cds5, and Cs2bi2mns5 Fu Qiang Huang, Rebecca C
ARTICLE IN PRESS Journal of Solid State Chemistry 174 (2003) 334–341 Syntheses, structures, optical properties, and theoretical calculations of Cs2Bi2ZnS5,Cs2Bi2CdS5, and Cs2Bi2MnS5 Fu Qiang Huang, Rebecca C. Somers, Adam D. McFarland, Richard P. Van Duyne, and James A Ibersà Department of Chemistry, Northwestern University, 2145 Sheridan Road, Evanston, IL 60208-3113, USA Received 24 December 2002; received in revised form 14 April 2003; accepted 25 April 2003 Abstract Three new compounds, Cs2Bi2ZnS5,Cs2Bi2CdS5, and Cs2Bi2MnS5, have been synthesized from the respective elements and a reactive flux Cs2S3 at 973 K. The compounds are isostructural and crystallize in a new structure type in space group Pnma of the orthorhombic system with four formula units in cells of dimensions at 153 K of a=15.763(3), b=4.0965(9), c=18.197(4) A˚ , ˚ 3 ˚ ˚ 3 V=1175.0(4) A for Cs2Bi2ZnS5; a=15.817(2), b=4.1782(6), c=18.473(3) A, V=1220.8(3) A for Cs2Bi2CdS5; and a=15.830(2), ˚ ˚ 3 2 2À b=4.1515(5), c=18.372(2) A, V=1207.4(2) A for Cs2Bi2MnS5. The structure is composed of two-dimensional N[Bi2MS5 ] (M=Zn, Cd, Mn) layers that stack perpendicular to the [100] axis and are separated by Cs+ cations. The layers consist of edge- 1 6À 1 4À sharing N[Bi2S6 ] and N[MS3 ] chains built from BiS6 octahedral and MS4 tetrahedral units. Two crystallographically unique Cs atoms are coordinated to S atoms in octahedral and monocapped trigonal prismatic environments. The structure of Cs2Bi2MS5,is 0 0 related to that of Na2ZrCu2S4 and those of the AMM Q3 materials (A=alkali metal, M=rare-earth or Group 4 element, M = Group 11 or 12 element, Q=chalcogen). -

Syllabus Outline
Syllabus Syllabus outline Syllabus component Recommended teaching hours SL HL Core 95 1. Stoichiometric relationships 13.5 2. Atomic structure 6 3. Periodicity 6 4. Chemical bonding and structure 13.5 5. Energetics/thermochemistry 9 6. Chemical kinetics 7 7. Equilibrium 4.5 8. Acids and bases 6.5 9. Redox processes 8 10. Organic chemistry 11 11. Measurement and data processing 10 Additional higher level (AHL) 60 12. Atomic structure 2 13. The periodic table—the transition metals 4 14. Chemical bonding and structure 7 15. Energetics/thermochemistry 7 16. Chemical kinetics 6 17. Equilibrium 4 18. Acids and bases 10 19. Redox processes 6 20. Organic chemistry 12 21. Measurement and analysis 2 Option 15 25 A. Materials 15 25 B. Biochemistry 15 25 C. Energy 15 25 D. Medicinal chemistry 15 25 20 Chemistry guide Syllabus outline Syllabus component Recommended teaching hours SL HL Practical scheme of work 40 60 Practical activities 20 40 Individual investigation (internal assessment—IA) 10 10 Group 4 project 10 10 Total teaching hours 150 240 The recommended teaching time is 240 hours to complete HL courses and 150 hours to complete SL courses as stated in the document General regulations: Diploma Programme (2011) (page 4, Article 8.2). Chemistry guide 21 Syllabus Approaches to the teaching of chemistry Format of the syllabus The format of the syllabus section of the group 4 guides is the same for each subject physics, chemistry and biology. This new structure gives prominence and focus to the teaching and learning aspects. Topics or options Topics are numbered and options are indicated by a letter. -

Macrocycles Containing 1, 1'-Ferrocenyldiselenolato Ligands On
Dalton Transactions View Article Online PAPER View Journal | View Issue Macrocycles containing 1,1’-ferrocenyldiselenolato ligands Cite this: Dalton Trans., 2018, 47, 5415 on group 4 metallocenes† Clara Sanchez-Perez,a,b Caroline E. Knapp, b Minna M. Karjalainen,a Raija Oilunkaniemi,a Claire J. Carmalt *b and Risto S. Laitinen *a 5 5 t Macrocyclic [Fe(η -C5H4Se)2M(η -C5H4R)2]2 [M = Ti (1), Zr (2), Hf (3), R = H; and M = Zr (4), Hf (5), R = Bu] were prepared and characterized by 77Se NMR spectroscopy and the crystal structures of 1–3 and 5 were determined by single-crystal X-ray diffraction. The crystal structure of 4 is known and the complex is iso- morphous with 5. 1–5 form mutually similar macrocyclic tetranuclear complexes in which the alternating Received 23rd January 2018, Fe(C5H4Se)2 and M(C5H4R)2 centers are linked by selenium bridges. The thermogravimetric analysis (TGA) Accepted 9th March 2018 of 1–3 under a helium atmosphere indicated that the complexes undergo a two-step decomposition DOI: 10.1039/c8dt00300a upon heating. The final products were identified using powder X-ray diffraction as FexMSe2, indicating Creative Commons Attribution-NonCommercial 3.0 Unported Licence. rsc.li/dalton their potential as single-source precursors for functional materials. Introduction been widely developed after the discovery that these materials may be used as precursors for semiconducting binary metal Transition metal dichalcogenides (TMDs) have been widely chalcogenides.13 Extension of this area to include various studied over the years due to their ability to intercalate species metallocene dichalcogenides has been possible because of the with apparent implications in catalytic and biological development of equipment for air-sensitive synthesis. -

Upper Limit of the Periodic Table and the Future Superheavy Elements
CLASSROOM Rajarshi Ghosh Upper Limit of the Periodic Table and the Future Department of Chemistry The University of Burdwan ∗ Superheavy Elements Burdwan 713 104, India. Email: [email protected] Controversy surrounds the isolation and stability of the fu- ture transactinoid elements (after oganesson) in the periodic table. A single conclusion has not yet been drawn for the highest possible atomic number, though there are several the- oretical as well as experimental results regarding this. In this article, the scientific backgrounds of those upcoming super- heavy elements (SHE) and their proposed electronic charac- ters are briefly described. Introduction Totally 118 elements, starting from hydrogen (atomic number 1) to oganesson (atomic number 118) are accommodated in the mod- ern form of the periodic table comprising seven periods and eigh- teen groups. Total 92 natural elements (if technetium is consid- ered as natural) are there in the periodic table (up to uranium hav- ing atomic number 92). In the actinoid series, only four elements— Keywords actinium, thorium, protactinium and uranium—are natural. The Superheavy elements, actinoid rest of the eleven elements—from neptunium (atomic number 93) series, transactinoid elements, periodic table. to lawrencium (atomic number 103)—are synthetic. Elements after actinoids (i.e., from rutherfordium) are called transactinoid elements. These are also called superheavy elements (SHE) as they have very high atomic numbers. Prof. G T Seaborg had Elements after actinoids a very distinct contribution in the field of transuranium element (i.e., from synthesis. For this, Prof. Seaborg was awarded the Nobel Prize in rutherfordium) are called transactinoid elements. 1951.