Title Analysis of the Genome Architecture of The
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Lineage-Specific Evolution of the Vertebrate Otopetrin Gene Family Revealed by Comparative Genomic Analyses
Hurle et al. BMC Evolutionary Biology 2011, 11:23 http://www.biomedcentral.com/1471-2148/11/23 RESEARCHARTICLE Open Access Lineage-specific evolution of the vertebrate Otopetrin gene family revealed by comparative genomic analyses Belen Hurle1, Tomas Marques-Bonet2,3, Francesca Antonacci3, Inna Hughes4, Joseph F Ryan1, NISC Comparative Sequencing Program1,5, Evan E Eichler3, David M Ornitz6, Eric D Green1,5* Abstract Background: Mutations in the Otopetrin 1 gene (Otop1) in mice and fish produce an unusual bilateral vestibular pathology that involves the absence of otoconia without hearing impairment. The encoded protein, Otop1, is the only functionally characterized member of the Otopetrin Domain Protein (ODP) family; the extended sequence and structural preservation of ODP proteins in metazoans suggest a conserved functional role. Here, we use the tools of sequence- and cytogenetic-based comparative genomics to study the Otop1 and the Otop2-Otop3 genes and to establish their genomic context in 25 vertebrates. We extend our evolutionary study to include the gene mutated in Usher syndrome (USH) subtype 1G (Ush1g), both because of the head-to-tail clustering of Ush1g with Otop2 and because Otop1 and Ush1g mutations result in inner ear phenotypes. Results: We established that OTOP1 is the boundary gene of an inversion polymorphism on human chromosome 4p16 that originated in the common human-chimpanzee lineage more than 6 million years ago. Other lineage- specific evolutionary events included a three-fold expansion of the Otop genes in Xenopus tropicalis and of Ush1g in teleostei fish. The tight physical linkage between Otop2 and Ush1g is conserved in all vertebrates. -

Fish and Fishery Products Hazards and Controls Guidance Fourth Edition – APRIL 2011
SGR 129 Fish and Fishery Products Hazards and Controls Guidance Fourth Edition – APRIL 2011 DEPARTMENT OF HEALTH AND HUMAN SERVICES PUBLIC HEALTH SERVICE FOOD AND DRUG ADMINISTRATION CENTER FOR FOOD SAFETY AND APPLIED NUTRITION OFFICE OF FOOD SAFETY Fish and Fishery Products Hazards and Controls Guidance Fourth Edition – April 2011 Additional copies may be purchased from: Florida Sea Grant IFAS - Extension Bookstore University of Florida P.O. Box 110011 Gainesville, FL 32611-0011 (800) 226-1764 Or www.ifasbooks.com Or you may download a copy from: http://www.fda.gov/FoodGuidances You may submit electronic or written comments regarding this guidance at any time. Submit electronic comments to http://www.regulations. gov. Submit written comments to the Division of Dockets Management (HFA-305), Food and Drug Administration, 5630 Fishers Lane, Rm. 1061, Rockville, MD 20852. All comments should be identified with the docket number listed in the notice of availability that publishes in the Federal Register. U.S. Department of Health and Human Services Food and Drug Administration Center for Food Safety and Applied Nutrition (240) 402-2300 April 2011 Table of Contents: Fish and Fishery Products Hazards and Controls Guidance • Guidance for the Industry: Fish and Fishery Products Hazards and Controls Guidance ................................ 1 • CHAPTER 1: General Information .......................................................................................................19 • CHAPTER 2: Conducting a Hazard Analysis and Developing a HACCP Plan -

Sequence Analysis and Comparative Study of the Protein Subunits of Archaeal Rnase P
biomolecules Review Sequence Analysis and Comparative Study of the Protein Subunits of Archaeal RNase P Manoj P. Samanta 1,*,†, Stella M. Lai 2,3,†, Charles J. Daniels 3,4 and Venkat Gopalan 2,3,* 1 Systemix Institute, Redmond, WA 98053, USA 2 Department of Chemistry & Biochemistry, The Ohio State University, Columbus, OH 43210, USA; [email protected] 3 Center for RNA Biology, The Ohio State University, Columbus, OH 43210, USA; [email protected] 4 Department of Microbiology, The Ohio State University, Columbus, OH 43210, USA * Correspondence: [email protected] (M.P.S.); [email protected] (V.G.); Tel.: +1-425-898-8187 (M.P.S.); +1-614-292-1332 (V.G.) † These authors contributed equally to this work. Academic Editor: Denis Drainas Received: 1 March 2016; Accepted: 8 April 2016; Published: 20 April 2016 Abstract: RNase P, a ribozyme-based ribonucleoprotein (RNP) complex that catalyzes tRNA 51-maturation, is ubiquitous in all domains of life, but the evolution of its protein components (RNase P proteins, RPPs) is not well understood. Archaeal RPPs may provide clues on how the complex evolved from an ancient ribozyme to an RNP with multiple archaeal and eukaryotic (homologous) RPPs, which are unrelated to the single bacterial RPP. Here, we analyzed the sequence and structure of archaeal RPPs from over 600 available genomes. All five RPPs are found in eight archaeal phyla, suggesting that these RPPs arose early in archaeal evolutionary history. The putative ancestral genomic loci of archaeal RPPs include genes encoding several members of ribosome, exosome, and proteasome complexes, which may indicate coevolution/coordinate regulation of RNase P with other core cellular machineries. -

Analysis of Gene Expression Data for Gene Ontology
ANALYSIS OF GENE EXPRESSION DATA FOR GENE ONTOLOGY BASED PROTEIN FUNCTION PREDICTION A Thesis Presented to The Graduate Faculty of The University of Akron In Partial Fulfillment of the Requirements for the Degree Master of Science Robert Daniel Macholan May 2011 ANALYSIS OF GENE EXPRESSION DATA FOR GENE ONTOLOGY BASED PROTEIN FUNCTION PREDICTION Robert Daniel Macholan Thesis Approved: Accepted: _______________________________ _______________________________ Advisor Department Chair Dr. Zhong-Hui Duan Dr. Chien-Chung Chan _______________________________ _______________________________ Committee Member Dean of the College Dr. Chien-Chung Chan Dr. Chand K. Midha _______________________________ _______________________________ Committee Member Dean of the Graduate School Dr. Yingcai Xiao Dr. George R. Newkome _______________________________ Date ii ABSTRACT A tremendous increase in genomic data has encouraged biologists to turn to bioinformatics in order to assist in its interpretation and processing. One of the present challenges that need to be overcome in order to understand this data more completely is the development of a reliable method to accurately predict the function of a protein from its genomic information. This study focuses on developing an effective algorithm for protein function prediction. The algorithm is based on proteins that have similar expression patterns. The similarity of the expression data is determined using a novel measure, the slope matrix. The slope matrix introduces a normalized method for the comparison of expression levels throughout a proteome. The algorithm is tested using real microarray gene expression data. Their functions are characterized using gene ontology annotations. The results of the case study indicate the protein function prediction algorithm developed is comparable to the prediction algorithms that are based on the annotations of homologous proteins. -

Molecular and Physiological Basis for Hair Loss in Near Naked Hairless and Oak Ridge Rhino-Like Mouse Models: Tracking the Role of the Hairless Gene
University of Tennessee, Knoxville TRACE: Tennessee Research and Creative Exchange Doctoral Dissertations Graduate School 5-2006 Molecular and Physiological Basis for Hair Loss in Near Naked Hairless and Oak Ridge Rhino-like Mouse Models: Tracking the Role of the Hairless Gene Yutao Liu University of Tennessee - Knoxville Follow this and additional works at: https://trace.tennessee.edu/utk_graddiss Part of the Life Sciences Commons Recommended Citation Liu, Yutao, "Molecular and Physiological Basis for Hair Loss in Near Naked Hairless and Oak Ridge Rhino- like Mouse Models: Tracking the Role of the Hairless Gene. " PhD diss., University of Tennessee, 2006. https://trace.tennessee.edu/utk_graddiss/1824 This Dissertation is brought to you for free and open access by the Graduate School at TRACE: Tennessee Research and Creative Exchange. It has been accepted for inclusion in Doctoral Dissertations by an authorized administrator of TRACE: Tennessee Research and Creative Exchange. For more information, please contact [email protected]. To the Graduate Council: I am submitting herewith a dissertation written by Yutao Liu entitled "Molecular and Physiological Basis for Hair Loss in Near Naked Hairless and Oak Ridge Rhino-like Mouse Models: Tracking the Role of the Hairless Gene." I have examined the final electronic copy of this dissertation for form and content and recommend that it be accepted in partial fulfillment of the requirements for the degree of Doctor of Philosophy, with a major in Life Sciences. Brynn H. Voy, Major Professor We have read this dissertation and recommend its acceptance: Naima Moustaid-Moussa, Yisong Wang, Rogert Hettich Accepted for the Council: Carolyn R. -

The Genome of Nanoarchaeum Equitans: Insights Into Early Archaeal Evolution and Derived Parasitism
The genome of Nanoarchaeum equitans: Insights into early archaeal evolution and derived parasitism Elizabeth Waters†‡, Michael J. Hohn§, Ivan Ahel¶, David E. Graham††, Mark D. Adams‡‡, Mary Barnstead‡‡, Karen Y. Beeson‡‡, Lisa Bibbs†, Randall Bolanos‡‡, Martin Keller†, Keith Kretz†, Xiaoying Lin‡‡, Eric Mathur†, Jingwei Ni‡‡, Mircea Podar†, Toby Richardson†, Granger G. Sutton‡‡, Melvin Simon†, Dieter So¨ ll¶§§¶¶, Karl O. Stetter†§¶¶, Jay M. Short†, and Michiel Noordewier†¶¶ †Diversa Corporation, 4955 Directors Place, San Diego, CA 92121; ‡Department of Biology, San Diego State University, 5500 Campanile Drive, San Diego, CA 92182; §Lehrstuhl fu¨r Mikrobiologie und Archaeenzentrum, Universita¨t Regensburg, Universita¨tsstrasse 31, D-93053 Regensburg, Germany; ‡‡Celera Genomics Rockville, 45 West Gude Drive, Rockville, MD 20850; Departments of ¶Molecular Biophysics and Biochemistry and §§Chemistry, Yale University, New Haven, CT 06520-8114; and ʈDepartment of Biochemistry, Virginia Polytechnic Institute and State University, Blacksburg, VA 24061 Communicated by Carl R. Woese, University of Illinois at Urbana–Champaign, Urbana, IL, August 21, 2003 (received for review July 22, 2003) The hyperthermophile Nanoarchaeum equitans is an obligate sym- (6–8). Genomic DNA was either digested with restriction en- biont growing in coculture with the crenarchaeon Ignicoccus. zymes or sheared to provide clonable fragments. Two plasmid Ribosomal protein and rRNA-based phylogenies place its branching libraries were made by subcloning randomly sheared fragments point early in the archaeal lineage, representing the new archaeal of this DNA into a high-copy number vector (Ϸ2.8 kbp library) kingdom Nanoarchaeota. The N. equitans genome (490,885 base or low-copy number vector (Ϸ6.3 kbp library). DNA sequence pairs) encodes the machinery for information processing and was obtained from both ends of plasmid inserts to create repair, but lacks genes for lipid, cofactor, amino acid, or nucleotide ‘‘mate-pairs,’’ pairs of reads from single clones that should be biosyntheses. -

Periodic and Coordinated Gene Expression Between a Diazotroph and Its Diatom Host
The ISME Journal (2019) 13:118–131 https://doi.org/10.1038/s41396-018-0262-2 ARTICLE Periodic and coordinated gene expression between a diazotroph and its diatom host 1 1,2 1 3 4 Matthew J. Harke ● Kyle R. Frischkorn ● Sheean T. Haley ● Frank O. Aylward ● Jonathan P. Zehr ● Sonya T. Dyhrman1,2 Received: 11 April 2018 / Revised: 28 June 2018 / Accepted: 28 July 2018 / Published online: 16 August 2018 © International Society for Microbial Ecology 2018 Abstract In the surface ocean, light fuels photosynthetic carbon fixation of phytoplankton, playing a critical role in ecosystem processes including carbon export to the deep sea. In oligotrophic oceans, diatom–diazotroph associations (DDAs) play a keystone role in ecosystem function because diazotrophs can provide otherwise scarce biologically available nitrogen to the diatom host, fueling growth and subsequent carbon sequestration. Despite their importance, relatively little is known about the nature of these associations in situ. Here we used metatranscriptomic sequencing of surface samples from the North Pacific Subtropical Gyre (NPSG) to reconstruct patterns of gene expression for the diazotrophic symbiont Richelia and we – 1234567890();,: 1234567890();,: examined how these patterns were integrated with those of the diatom host over day night transitions. Richelia exhibited significant diel signals for genes related to photosynthesis, N2 fixation, and resource acquisition, among other processes. N2 fixation genes were significantly co-expressed with host nitrogen uptake and metabolism, as well as potential genes involved in carbon transport, which may underpin the exchange of nitrogen and carbon within this association. Patterns of expression suggested cell division was integrated between the host and symbiont across the diel cycle. -

Molecular Interaction Between Fish Pathogens and Host Aquatic Animals
K. Tsukamoto, T. Kawamura, T. Takeuchi, T. D. Beard, Jr. and M. J. Kaiser, eds. Fisheries for Global Welfare and Environment, 5th World Fisheries Congress 2008, pp. 277–288. © by TERRAPUB 2008. Molecular Interaction between Fish Pathogens and Host Aquatic Animals Laura L. Brown* and Stewart C. Johnson National Research Council of Canada Institute for Marine Biosciences 1411 Oxford Street Halifax, NS, B3H 3Z1, Canada Present address: Fisheries and Oceans Canada Pacific Biological Station 3190 Hammond Bay Road Nanaimo, NS, V9T 6N7, Canada *E-mail: [email protected] We have studied the host-pathogen interactions between Atlantic salmon (Salmo salar L.) and Aeromonas salmonicida. Sequencing the genome of the bacterium allowed us to investigate virulence factors and other gene products with potential as vaccines. Using knock-out mutants of A. salmonicida, we identified key virulence factors. Proteomics studies of bacterial cells grown in a variety of media as well as in an in vivo implant system revealed differential protein production and have shed new light on bacterial proteins such as superoxide dismutase, pili and flagellar proteins, type three secretion systems, and their roles in A. salmonicida pathogenicity. We constructed a whole ge- nome DNA microarray to use in comparative genomic hybridizations (M-CGH) and bacterial gene expression studies. Carbohydrate analysis has shown the variation in LPS between strains and reveals the importance of LPS in viru- lence. Salmon were challenged with A. salmonicida and tissues were taken to construct suppressive subtractive hybridization libraries to investigate differ- ential host gene expression. We constructed an Atlantic salmon cDNA microarray to investigate the host response to A. -

RNA Epigenetics: Fine-Tuning Chromatin Plasticity and Transcriptional Regulation, and the Implications in Human Diseases
G C A T T A C G G C A T genes Review RNA Epigenetics: Fine-Tuning Chromatin Plasticity and Transcriptional Regulation, and the Implications in Human Diseases Amber Willbanks, Shaun Wood and Jason X. Cheng * Department of Pathology, Hematopathology Section, University of Chicago, Chicago, IL 60637, USA; [email protected] (A.W.); [email protected] (S.W.) * Correspondence: [email protected] Abstract: Chromatin structure plays an essential role in eukaryotic gene expression and cell identity. Traditionally, DNA and histone modifications have been the focus of chromatin regulation; however, recent molecular and imaging studies have revealed an intimate connection between RNA epigenetics and chromatin structure. Accumulating evidence suggests that RNA serves as the interplay between chromatin and the transcription and splicing machineries within the cell. Additionally, epigenetic modifications of nascent RNAs fine-tune these interactions to regulate gene expression at the co- and post-transcriptional levels in normal cell development and human diseases. This review will provide an overview of recent advances in the emerging field of RNA epigenetics, specifically the role of RNA modifications and RNA modifying proteins in chromatin remodeling, transcription activation and RNA processing, as well as translational implications in human diseases. Keywords: 5’ cap (5’ cap); 7-methylguanosine (m7G); R-loops; N6-methyladenosine (m6A); RNA editing; A-to-I; C-to-U; 2’-O-methylation (Nm); 5-methylcytosine (m5C); NOL1/NOP2/sun domain Citation: Willbanks, A.; Wood, S.; (NSUN); MYC Cheng, J.X. RNA Epigenetics: Fine-Tuning Chromatin Plasticity and Transcriptional Regulation, and the Implications in Human Diseases. Genes 2021, 12, 627. -
Primepcr™Assay Validation Report
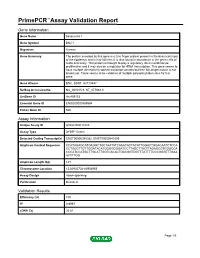
PrimePCR™Assay Validation Report Gene Information Gene Name basonuclin 1 Gene Symbol BNC1 Organism Human Gene Summary The protein encoded by this gene is a zinc finger protein present in the basal cell layer of the epidermis and in hair follicles. It is also found in abundance in the germ cells of testis and ovary. This protein is thought to play a regulatory role in keratinocyte proliferation and it may also be a regulator for rRNA transcription. This gene seems to have multiple alternatively spliced transcript variants but their full-length nature is not known yet. There seems to be evidence of multiple polyadenylation sites for this gene. Gene Aliases BNC, BSN1, HsT19447 RefSeq Accession No. NC_000015.9, NT_077661.3 UniGene ID Hs.459153 Ensembl Gene ID ENSG00000169594 Entrez Gene ID 646 Assay Information Unique Assay ID qHsaCID0017223 Assay Type SYBR® Green Detected Coding Transcript(s) ENST00000345382, ENST00000541809 Amplicon Context Sequence CCATAGAGCATGAGGCTGCTAATATCAAACACTACATTGGACTGGACAATCTCCA CCTGGCTTGTTGGATACATGGGGGGGATCCTTAGCTTACTTAGAGCGTGGGCCA CCCATCCATGCTTGCATTGGTCACACTGACGGTGGTTTATTTTCCCGGGTTTGAA ACTTTGG Amplicon Length (bp) 141 Chromosome Location 15:83935724-83936955 Assay Design Intron-spanning Purification Desalted Validation Results Efficiency (%) 100 R2 0.9997 cDNA Cq 26.81 Page 1/5 PrimePCR™Assay Validation Report cDNA Tm (Celsius) 84.5 gDNA Cq 39.42 Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 2/5 PrimePCR™Assay Validation Report BNC1, Human Amplification -

The Mínimum Cell
The minimum cell GENOMIC – ADVANCED GENETICS AUTHOR: I. ODEI BARREÑADA Overview The Concept Small genomes Minimal gene set Approache theories Minimal genome proyect Future insight The concept “MINIMUN CELL“ The smallest size (of genetic The smallest unit of life that can information) replicate autonomously = minimum genome Small genomes Circovirus (1.800 base pairs / 3 gens) Virus Carsonella ruddi (159 kb /182 genes) Symbiont Nanoarchaeum equitans (490 kb/ 553 genes) Parasite Mycoplasma genitalium (582 kb/ 521 genes ) Parasite Pelagibacter ubique (1,3 Mb/ 1,370 genes) Free-living Smallest genomes Circovirus (1.800 base pairs / 3 gens) Virus Carsonella ruddi (159 kb /182 genes) Symbiont Nanoarchaeum equitans (490 kb/ 553 genes) Parasite Mycoplasma genitalium (582 kb/ 521 genes ) Parasite Pelagibacter ubique (1,3 Mb/ 1,370 genes) Free-living (Giovannoni et al., 2005) “MINIMAL GENE SET” By genome comparison Ubiquitous genes: Translation Transcription Replication of DNA Variable genes: Depends of environment (Koonin, 2003) Theories for reach the minimum cell Two approaches Top – Down knock-down known organism Bottom-up --> build from synthetics DNA Minimal genome project J. Craig Venter Institute Search of essential genes Gene disruption by transposons Seq the survivors organisms Detect the disrupted gene Declare this genes as NON-ESENTIAL In M. genitalium only 382 of 521 genes are essential (Gibson et al., 2011) Build de novo M. genitalium genome Future insight - Cell design Create artificial cells for: Generation of hydrogen for fuel Capturing excess carbon dioxide in the atmosphere Drug delivery directly into the body As Enzyme therapy Artificial blood cells … And all you can think References Koonin E V. -

Proteomics Provides Insights Into the Inhibition of Chinese Hamster V79
www.nature.com/scientificreports OPEN Proteomics provides insights into the inhibition of Chinese hamster V79 cell proliferation in the deep underground environment Jifeng Liu1,2, Tengfei Ma1,2, Mingzhong Gao3, Yilin Liu4, Jun Liu1, Shichao Wang2, Yike Xie2, Ling Wang2, Juan Cheng2, Shixi Liu1*, Jian Zou1,2*, Jiang Wu2, Weimin Li2 & Heping Xie2,3,5 As resources in the shallow depths of the earth exhausted, people will spend extended periods of time in the deep underground space. However, little is known about the deep underground environment afecting the health of organisms. Hence, we established both deep underground laboratory (DUGL) and above ground laboratory (AGL) to investigate the efect of environmental factors on organisms. Six environmental parameters were monitored in the DUGL and AGL. Growth curves were recorded and tandem mass tag (TMT) proteomics analysis were performed to explore the proliferative ability and diferentially abundant proteins (DAPs) in V79 cells (a cell line widely used in biological study in DUGLs) cultured in the DUGL and AGL. Parallel Reaction Monitoring was conducted to verify the TMT results. γ ray dose rate showed the most detectable diference between the two laboratories, whereby γ ray dose rate was signifcantly lower in the DUGL compared to the AGL. V79 cell proliferation was slower in the DUGL. Quantitative proteomics detected 980 DAPs (absolute fold change ≥ 1.2, p < 0.05) between V79 cells cultured in the DUGL and AGL. Of these, 576 proteins were up-regulated and 404 proteins were down-regulated in V79 cells cultured in the DUGL. KEGG pathway analysis revealed that seven pathways (e.g.