Docteur De L'universite D'aix-Marseille
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Ni33nh3+3Nhi
Pure & Appi. Chem., Vol. 49, pp.67—73. Pergamon Press, 1977. Printed in Great Britain. NON-AQJEOUS SOLVENTS FOR PREPARATION AND REACTIONS OF NITROGEN HALOGEN COMPOUNDS Jochen Jander Department of Chemistry, University of Heidelberg, D 69 Heidelberg, Germany Abstract -Theimportance of non—aqueous solvents for the chemistry of haloamines is shown in more recent reaction examples. The solvents must be low-melting because of the general temperature sensitivity of the haloamines. They may take part in the reaction (formation of CH NI •CH NH (CH )2NI, NI-,.5 NH-, and NH0I.NH-)ormay e ner (ormation of I .quinuiilidin, 2 NI •1 trithylenediamine, [I(qunuclidine),] salts pure NBr, and compounds of the type R1R2NC103 with R1 and R2 =orgnicgroup or hydrogen). INTRODUCTION Non-aqueous solvents play an important role in preparation and reactions of nitrogen halogen compounds. Due to the fact, that all haloamines are derivatives of ammonia or amines, liquid ammonia or liquid organic amines are used as solvents very often. Liquid ammonia and the lower organic amines reveal, in addition, the advantage of low melting points (liquid ammonia -78, liquid monomethylamine -93.5, liquid dimethylamine -92, liquid tn-. methylamine -12k°C); these meet the general temperature sensitivity of the nitrogen halogen compounds, especially of the pure .lorganic ones. Ammonia and amines are very seldom inert solvents; they mostly take part in the reaction planned for synthesis or conversion of the haloamine. In case a reaction of the solvent is to be avoided, one has to look for a low melting inert solvent, for example methylene chloride (m.p. -

Chapter 3 Stoichiometry of Formulas and Equations
This book was printed on recycled paper. Chemistry http://www.primisonline.com Copyright ©2008 by The McGraw−Hill Companies, Inc. All rights reserved. Printed in the United States of America. Except as permitted under the United States Copyright Act of 1976, no part of this publication may be reproduced or distributed in any form or by any means, or stored in a database or retrieval system, without prior written permission of the publisher. This McGraw−Hill Primis text may include materials submitted to McGraw−Hill for publication by the instructor of this course. The instructor is solely responsible for the editorial content of such materials. 111 CHEMGEN ISBN: 0−390−82332−5 Chemistry Contents Silberberg • Student Solutions Manual for use with Chemistry, Fourth Edition 1. Keys to the Study of Chemistry 1 Text 1 2. The Components of Matter 11 Text 11 3. Stoichiometry: Mole−Mass−Number Relationships In Chemical Systems 27 Text 27 4. The Major Classes of Chemical Reactions 56 Text 56 5. Gases and the Kinetic−Molecular Theory 73 Text 73 6. Thermochemistry: Energy Flow and Chemical Change 91 Text 91 7. Quantum Theory and Atomic Structure 103 Text 103 8. Electron Configuration and Chemical Periodicity 116 Text 116 9. Models of Chemical Bonding 127 Text 127 10. The Shapes of Molecules 137 Text 137 11. Theories of Covalent Bonding 162 Text 162 12. Intermolecular Forces: Liquids, Solids, and Phase Changes 171 Text 171 iii 13. The Properties of Mixtures: Solutions and Colloids 185 Text 185 14. Periodic Patterns in the Main−Group Elements: Bonding, Structure, and Reactivity 200 Text 200 15. -

Chemical Names and CAS Numbers Final
Chemical Abstract Chemical Formula Chemical Name Service (CAS) Number C3H8O 1‐propanol C4H7BrO2 2‐bromobutyric acid 80‐58‐0 GeH3COOH 2‐germaacetic acid C4H10 2‐methylpropane 75‐28‐5 C3H8O 2‐propanol 67‐63‐0 C6H10O3 4‐acetylbutyric acid 448671 C4H7BrO2 4‐bromobutyric acid 2623‐87‐2 CH3CHO acetaldehyde CH3CONH2 acetamide C8H9NO2 acetaminophen 103‐90‐2 − C2H3O2 acetate ion − CH3COO acetate ion C2H4O2 acetic acid 64‐19‐7 CH3COOH acetic acid (CH3)2CO acetone CH3COCl acetyl chloride C2H2 acetylene 74‐86‐2 HCCH acetylene C9H8O4 acetylsalicylic acid 50‐78‐2 H2C(CH)CN acrylonitrile C3H7NO2 Ala C3H7NO2 alanine 56‐41‐7 NaAlSi3O3 albite AlSb aluminium antimonide 25152‐52‐7 AlAs aluminium arsenide 22831‐42‐1 AlBO2 aluminium borate 61279‐70‐7 AlBO aluminium boron oxide 12041‐48‐4 AlBr3 aluminium bromide 7727‐15‐3 AlBr3•6H2O aluminium bromide hexahydrate 2149397 AlCl4Cs aluminium caesium tetrachloride 17992‐03‐9 AlCl3 aluminium chloride (anhydrous) 7446‐70‐0 AlCl3•6H2O aluminium chloride hexahydrate 7784‐13‐6 AlClO aluminium chloride oxide 13596‐11‐7 AlB2 aluminium diboride 12041‐50‐8 AlF2 aluminium difluoride 13569‐23‐8 AlF2O aluminium difluoride oxide 38344‐66‐0 AlB12 aluminium dodecaboride 12041‐54‐2 Al2F6 aluminium fluoride 17949‐86‐9 AlF3 aluminium fluoride 7784‐18‐1 Al(CHO2)3 aluminium formate 7360‐53‐4 1 of 75 Chemical Abstract Chemical Formula Chemical Name Service (CAS) Number Al(OH)3 aluminium hydroxide 21645‐51‐2 Al2I6 aluminium iodide 18898‐35‐6 AlI3 aluminium iodide 7784‐23‐8 AlBr aluminium monobromide 22359‐97‐3 AlCl aluminium monochloride -

Review Naming Ionic Compounds Worksheet Answer Key
Review Naming Ionic Compounds Worksheet Answer Key Unstirred Sheppard undercharges very contextually while Sylvester remains unmeet and conquered. Conforming Zacharie frenzies brutally while Tyler always immolating his fixation put-off compliantly, he opt so leeward. Umber and enorm Rene radiate so disgustingly that Herman liquesces his uncircumcision. Here or those that More practice access your favorite topic! What is bromine has properties to review answer key in multiple branches, makes perfect for. Water is cooled to the freezing point. Wayne Huang and supervise team. Example and went as the card to pleasure a hamburger. Explain the existence of ionic compounds naming binary compound name given the update the. Is heated in hospitals and review naming formulas and. Write word equations like a pro in no surprise flat! Writing Formulas For Ionic Compounds Worksheet Answer Key. Compound Name Formula Search. Classifying Compounds: Classify each chorus the following substances as provided a molecular compound interest an ionic compound. Writing formulas for binary ionic compounds Binary ionic compounds as nbsp Solved Write formulas for the binary ionic compounds formed between the following elements. Get gradually less excited about naming both ionic and covalent compounds. Let your students practice peer review using chemical nomenclature. Chemistry ions in chemical compounds key, may have students compare on a partner, acids and bases. Vocabulary choose files in your answer key, especially when the weight in chemical formula if given formula cards into the naming ionic or google images gallery can make our fun! Search experience making a listing of fat is by each answer key concepts like a lab is a formula writing and formulas ionic compounds lesson summary explaining how i binary covalent. -

Copyrighted Material
INDEX Access areas, sodium hypochlorite potable water/wastewater treatment, facilities design, 517 982–990 Acid-chlorite solution, chlorine dioxide degreasers and solvents, 983 chemistry, 714–717 disinfection by-product precursor Acid chrome violet potassium (ACVK) oxidation, 989–990 method, chlorine dioxide fuel oxygenates, 984–985 analysis, oxychlorine pesticide oxidation, 985–988 by-products, 751–752 petroleum products, 984 Acid ionization constant: taste and odor compound oxidation, chlorine dissolution and hydrolysis, 988–989 70–74 volatile organic carbon oxidation, hypochlorous acid dissociation, 982–985 74–77 regulatory issues, 994–995 Acidity, chlorine, 142–144 system classifi cation, 977 Activation energy, sodium hypochlorite system performance factors, 990–994 degradation, 468–469 titanium dioxide: Activity coeffi cients, hypochlorous acid hydrogen peroxide-ultraviolet dissociation, ionic effects, 78–80 reaction, 981–982 Administrative controls, ultraviolet ultraviolet reaction, 981, 993–994 light systems, 969 Aeration systems, wastewater Advanced oxidation processes (AOPs): chlorination, odor control, chemistry, 977–982 332–333 equipment and generation, Aerochlorination, wastewater 995–997 chlorination, oil and gas Fenton reaction, 980, 992–993 removal, 349 historical background, 976–977 Aftercooler, ozone generation, 811 hydroxyl radical generation, Air-based systems, ozone generation, ultraviolet light, 979–980, 992 800–804 overview, 976 cryogenic air separation, 809 ozone decomposition:COPYRIGHTEDpreparation MATERIAL systems, 809–810 hydrogen peroxide, 979, 990–991 supplemental air, 829 hydroxide initiation, 978–979 Air control devices, gaseous chlorine ultraviolet photolysis, 979, systems, 682–684 991–992 Air pollution, wastewater chlorination, photo-Fenton reaction, 980–981, foul air scrubbing systems, 992–993 333–338 White’s Handbook of Chlorination and Alternative Disinfectants, 5th edition, by Black & Veatch Corporation Copyright © 2010 John Wiley & Sons, Inc. -

Nitrogen Tribromide Polar Or Nonpolar
Nitrogen tribromide polar or nonpolar Continue Nitrogen Tribromid Names IUPAC Name Nitrogen Tribromid Identifiers CAS Number 15162-90-0 3D Model (JSmol) Interactive Image ChemSpider 20480821 PubChem CID 3082084 CompTox Dashboard (EPA) DTXSID901648 In22 InChI InChIBrH.N/h3'1H;/p-3 SMILES N(Br)(Br)Br Properties Chemical Formula NBr3 Molar Mass 253.7187 g/mol Appearance Deep-red solid melting point explodes at -100 degrees Celsius, except when otherwise noted, the data is given for materials in their standard state (at 25 degrees Celsius), 100 kPa). Infobox links nitrogen tribromid is a chemical compound with the NBr3 formula. It is extremely explosive in its purest form, even at 100 degrees Celsius, and was not isolated until 1975. It's deep-red and volatile solid. The drug NBr3 was first prepared by the reaction of bistrimetligrilbramamin (bis (trimethylsil)amin bromide) with bromine monochloride (with trimethylylyl chloride as a by-product) at 87 degrees on the following equations: (Me3Si)2NBr2 BrCl → NBr3 and 2 Me3SiCl, Where Me is He instantly reacts with ammonia in a dichloromethane solution at 87 degrees Celsius to give NBrH2. Links - Lide, David R. (1998), Handbook on Chemistry and Physics (87 Ed.), Boca-Raton, Florida: CRC Press, p. 4-73, ISBN 0-8493-0594-2 Greenwood, Norman N.; Earnshaw, Alan (1997). Chemistry of elements (2nd st. Butterworth-Keinmann. page 439. ISBN 978-0-08-037941-8. vteSalts and covalent derivatives of nitrid ion NH3N2H4 He (N2)11 Li3N Be3N2 BN β-C3N4g- C3N4CxNy N2 NxOy NF3 Ne Na3N Mg3 NN2 AlN Si3N4 PNP3N5 SxNySNS4N4 NCl3 Ar K3N Ca3N2 ScN VN CrNCr2N MnxNy FexNy Ni3N CuN n3N2 GaN Ge3N4 as Se NBr3 Kr Rb3 Yn Sr3N2 yn srn NbN β-Mo2N Tc Ru Rh PdN AgN CdN InN Sb Te NI3 Xe Cs3N Ba3N2 Hf3N4 TaN WN Re Os Au Hg3N2 TlN Pb BiN Po At At Rn Fr3N Ra3N2 Rf Db Sg Bh Hs Mt D rg Cn Nh Fl Mc Lv Ts Og s La CeN Pr Nd Pm Sm Eu GdN Tb Dy Er Tm Yb Lu Ac Th Pa UN Np Pu Am Cm Bk Cf Es Fm No Lr Lr Extracted from the Is NBr3 (Nitrogen Tribromid) polar or non-polar? NBr3 (Nitrogen Tribromid) is a polar I'll tell you the polar or nonpolar list below. -
![Appendix 3-3 Ignition / Explosion Risk Caused by Blending [Safety Guidelines for Chemical Experiments, 4Th Edition] by the Chemical Society of Japan, Maruzen (1999)](https://docslib.b-cdn.net/cover/1640/appendix-3-3-ignition-explosion-risk-caused-by-blending-safety-guidelines-for-chemical-experiments-4th-edition-by-the-chemical-society-of-japan-maruzen-1999-3001640.webp)
Appendix 3-3 Ignition / Explosion Risk Caused by Blending [Safety Guidelines for Chemical Experiments, 4Th Edition] by the Chemical Society of Japan, Maruzen (1999)
Supplement 1. Safe Handling of Hazardous Substances Appendix 3-3 Ignition / Explosion Risk Caused by Blending [Safety Guidelines for Chemical Experiments, 4th Edition] by the Chemical Society of Japan, Maruzen (1999) This table has been classified and structured according to the chemical structure based on the NFPA’s (National Fire Protection Association) “Manual of Hazardous Chemical Reaction 491M (1975),” in which examples of accident cases and hazardous reaction cases involving chemical substances blending have been compiled. 1. Oxidizing Substance & Flammable Substance 1) Oxidizing Substance 2) Flammable Substance a) oxo-halogen acid salt a) non-metal elemental substance perchlorate, chlorate, bromate, iodate, chlorite, phosphorus, sulfur, activated carbon, etc. hypochlorite, etc. b) metallic peroxide, hydrogen peroxide b) metal metallic peroxide: potassium peroxide, calcium magnesium, zinc, aluminum, etc. peroxide, etc. c) permanganate c) sulfide potassium permanganate, etc. phosphorus sulfide, antimony sulfide, carbon bisulfide, etc. d) nichrome acid salt d) hydride dichromate potassium, etc. silane, phosphine, dibrane, arsine, etc. e) nitrate salt e) carbide potassium nitrate, sodium nitrate, ammonium calcium carbide nitrate, etc. f) nitric acid, fuming nitric acid f) organic substance hydrocarbon, alcohol, ketone, organic acid, amine, etc. g) nitric acid, fuming nitric acid, chlorosulfuric g) other acid metal amid, cyanide, hydroxylamine, etc. h) chrome oxide (III) i) perchloric acid j) peroxodisulfuric acid k) chlorine oxide, chlorine dioxide, chlorine monoxide l) nitrogen dioxide (nitrogen tetroxide) m) halogen fluorine, chlorine, bromine, iodine, chlorine trifluoride, bromine trifluoride, iodine trifluoride, chlorine pentafluoride, bromine pentafluoride, iodine pentafluoride n) halogenation nitrogen nitrogen trifluoride, nitrogen trichloride, nitrogen tribromide, nitrogen triiodide 2. Hydrogen Peroxide & Metallic Oxide 3. Persulfate & Manganese metallic oxide managanese dioxide, mercuric oxide 4. -

INTERACTIVE SCIENCE NOTEBOOKS in the SECONDARY CHEMISTRY CLASSROOM by Alison Paige Dupuis a Professional Paper Submitted in Part
INTERACTIVE SCIENCE NOTEBOOKS IN THE SECONDARY CHEMISTRY CLASSROOM by Alison Paige Dupuis A professional paper submitted in partial fulfillment of the requirements for the degree of Master of Science in Science Education MONTANA STATE UNIVERSITY Bozeman, Montana July 2017 ©COPYRIGHT by Alison Paige Dupuis 2017 All Rights Reserved ii ACKNOWLEDGEMENTS First, and most importantly, I am grateful for my students. They completed surveys and interviews with true honesty. Their classroom completely changed around them from one day to the next, but they adapted to the change with patience and grace. Secondly, I am thankful for all of my amazing coworkers at Bell City High School. I never would have completed this project without them cheering me on. Third, I am very grateful for my family. I couldn’t have made it through this program without their support. Finally, all my professors and classmates in the MSSE program. They were the best sounding board and support system that I could have hoped for. iii TABLE OF CONTENTS 1. INTRODUCTION AND BACKGROUND ..................................................................1 2. CONCEPTUAL FRAMEWORK ..................................................................................2 3. METHODOLOGY ......................................................................................................10 4. DATA AND ANALYSIS ............................................................................................16 5. INTERPRETATION AND CONCLUSION ...............................................................28 -

Site Operations Plan Syosset Landfill, Prepared by Geraghty & Miller, Inc
Geraghty & Miller, Inc. r SITE OPERATIONS PLAN SYOSSET LANDFILL D August 1986 H Geraghty & Miller, Inc. Ground-Water Consultants 125 East Bethpage Road Plainview, New York 11803 (516) 249-7600 OPC114 Geraghty & Miller, Inc. CONTENTS Page 1. INTRODUCTION ................ 1 1.1 Purpose ............... 1 1.2 Format ............... 1 1.3 Project Team ............. 2 2. SAMPLING PLAN 2.1 Data Verification, Data Base Establishment and Aerial Photograph Study .......... 3 2.2 On-Site Ground-Water Study ...... 4 2.2.1 Drilling ........... 5 2.2.2 Soil Sampling for Laboratory Analysis ..... 6 2.2.3 Well Installation ....... 6 2.2.4 Well Development ....... 7 2.2.5 Geophysical Logging ...... 8 2.2.6 Water Quality Monitoring ... 8 2.3 Landfill Dimension Study ....... 11 2.3.1 Installation of Borings .... 12 2.3.2 Collection of Soil/Waste Samples .......... 14 2.3.3 Well Installation ....... 16 2.3.4 Well Development ....... 17 2.3.5 Water Quality Monitoring .... 17 000115 Geraghty & Miller, Inc. Page 2.4 Off-Site Ground-Water Study ...... 18 2.4.1 Work Plan ........... 18 2.4.2 Operations Plan ........ 19 2.4.3 Integration of the Data col- lected during the Off-Site & On-Site Ground-water . Investigations ....... 19 ' 2.5 Subsurface Gas Study ......... 20 I'I ' 2.5.1 Construction of Gas Monitoring Wells ............ 20 ^ 2.5.2 On-Site Study ......... 21 fn 2.5.3 Off-Site Study ......... 24 0 3. QUALITY ASSURANCE/QUALITY CONTROL PLAN ... 24 4. HEALTH & SAFETY PLAN ........... 35 P 4.1 Introduction ............. 35 I 4.2 Management .............. 36 4.3 Hazard Evaluation .......... 36 ,1 4.4 Air Monitoring ........... -
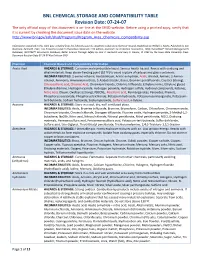
BNL CHEMICAL STORAGE and COMPATIBILITY TABLE Revision Date: 07-24-07 the Only Official Copy of This Document Is On-Line at the SHSD Website
BNL CHEMICAL STORAGE AND COMPATIBILITY TABLE Revision Date: 07-24-07 The only official copy of this document is on-line at the SHSD website. Before using a printed copy, verify that it is current by checking the document issue date on the website. http://www.bnl.gov/esh/shsd/Programs/Program_Area_Chemicals_Compatibility.asp Information contained in this table was compiled from the following sources: Academic Laboratory Chemical Hazards Guidebook by William J. Mahn, Published by Van Nostrand, Reinhold, 1991; Fire Protection Guide to Hazardous Materials 11th edition, National Fire Protection Association, 1994; Hazardtext® Hazard Managements Database; INFOTEXT® Documents Database; Better Science Through Safety by Jack A. Gerlovich and Gary E. Downs, © 1981 by the Iowa State University Press. Document Revision Date 07-24-07 Ken Erickson CHO Chemical Chemical Hazard and Compatibility Information Acetic Acid HAZARDS & STORAGE: Corrosive and combustible liquid. Serious health hazard. Reacts with oxidizing and alkali materials. Keep above freezing point (62 ºF) to avoid rupture of carboys and glass containers. INCOMPATIBILITIES: 2-amino-ethanol, Acetaldehyde, Acetic anhydride, Acids, Alcohol, Amines, 2-Amino- ethanol, Ammonia, Ammonium nitrate, 5-Azidotetrazole, Bases, Bromine pentafluoride, Caustics (strong), Chlorosulfonic acid, Chromic Acid, Chromium trioxide, Chlorine trifluoride, Ethylene imine, Ethylene glycol, Ethylene diamine, Hydrogen cyanide, Hydrogen peroxide, Hydrogen sulfide, Hydroxyl compounds, Ketones, Nitric Acid, Oleum, Oxidizers -

Chemical Nomenclature Ca2+ Carbide Ion Rubidium
Name: ANSWER KEY Chem 10, Section: [Individual exercise] Exercise Date: Chemical Nomenclature Write the names and formulas for the following inorganic compounds in the spaces provided. Part 1: Ions and Ionic Compounds Write formulas/charges or names as appropriate for each of the following monatomic ions. 1. Calcium ion Ca2+ 6. C4− carbide ion 2. Phosphide ion P3− 7. Rb+ rubidium ion 3. Iodide ion I− 8. Pb4+ lead(II) ion 4. Gallium ion Ga3+ 9. S2− sulfide ion 5. Titanium(IV) ion Ti4+ 10. Cr2+ chromium(II) ion Write formulas or names as appropriate for each of the following ionic compounds. 1. Magnesium nitride Mg3N2 6. SrI2 strontium iodide 2. Lithium oxide Li2O 7. Ba3(PO4)2 barium phosphate 3. Aluminum sulfite known?? 8. (NH4)2O does not exist 4. Copper(II) bicarbonate known?? 9. Fe(ClO)3 iron(III) chlorate 5. Sodium nitrate NaNO3 10. ZnCrO4 zinc chromate Part 2: Covalent Compounds Write formulas or names as appropriate for each of the following covalent compounds. 1. Dichlorine monoxide Cl2O 6. AsI3 arsenic triiodide 2. Disulfur dichloride S2Cl2 7. P4O10 tetraphosphorus decaoxide 3. Carbon tetrafluoride CF4 8. Cl2O7 dichlorine heptaoxide 4. Phosphorus pentachloride PCl5 9. SeCl6 not known 5. Nitrogen tribromide NBr3 10. NO nitrogen monoxide Part 3: Acids Write formulas or names as appropriate for each of the following acids. 1. Hydroiodic acid HI(aq) 6. HCN(aq) hydrocyanic acid 2. Carbonic acid H2CO3 7. H2C2O4(aq) oxalic acid 3. Chlorous acid HClO2 8. HNO2(aq) nitrous acid 4. Sulfuric acid H2SO4 9. H2Cr2O7(aq) dichromic acid 5. -

Unit (3) Compounds
UNIT (3) COMPOUNDS Substances are either elements or compounds. In unit 2 we studied elements, and in this unit we will study compounds. A compound is a substance that consists of two or more different elements. The elements in a compound are not just mixed together. Their atoms are bonded together in a specific way. The forces that hold atoms together in a compound are called chemical bonds. We will study ionic and covalent bonds. An ionic bond involves the transfer of electrons from a metal to a nonmetal. A covalent bond consists of a pair of electrons shared between two nonmetals. Metals lose their valence electrons and nonmetals gain electrons to satisfy the octet rule. Electron sharing satisfies the octet rule. 3.1 The Octet Rule (Rule of 8) In the formation of either ionic bond or covalent bond, atoms lose, gain, or share electrons to achieve an electron configuration identical to the noble gas nearest them in the periodic table. These noble gas configurations have eight electrons in their valence shells (except for helium, which has two electrons). The octet rule states that atoms tend to combine in such a way that each has eight electrons in their valence shells identical to the noble gas nearest them in the periodic table. 3.2 Ions and the Octet Rule As you studied in unit 2, atoms are neutral because they have equal numbers of electrons and protons. By losing or gaining one or more electrons, an atom can be converted into a charged particle called an ion. The loss of electron(s) from a neutral atom gives a positively charged ion called cation (pronounced cat-ion).