The Impact of Sleep Deprivation on Neuronal and Glial Signaling Pathways Important for Memory and Synaptic Plasticity
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Piccoli Zecchini Crescono Robert J
c, p •H<n j(|Mi ,>IV"."/A<; . vw*-» 1 -vT« . ^*^W •.\ WT*»«.*. •"**:'•> -: y***" «.tX»* »!»*'• tv* VVrrf^fti ' «-Vr . ... .. r ft - * irr^V^-^-W :*%"•£>-" ,*£% ^'£ZgL~$IS&XS>>«~t f^^lS^^^^t^^m^^..nW^S^JS**^S^**** :/ .'•. iSTOF^JT^Wf''.', **«».' *«*«« < ;»»«»r*tf* re f. i W-irt- « ** .T- . ,, , L'UNITÀ / GIOVEDÌ' 18 NOVEMBRE 1982 Fa scandalo il MILANO — Dopo aver strappato dal ventre umido e malsano (si / critici di- ili (iian Luigi Komli, -Il 12 L'epoca d'oro di Sanremo: la Fonit dice) degli archivi e nastroteche di Stato, i tesori della canzone mercato cinematografico- di •antica», la Fonit Cetra, unica casa fonografica del settore pubbli Umberto Itossi; -Il gruppo ci manifesto prosegue nella collana dei ricordi. co, è giunta agli anni che sono di Coppi, della Pizzi, di Tamhroni e di cinema nematografico pubbliio- ili di Nunzio Filogamo. Urlino torri; -I festival» ili di Antonioni? Ma Nilla Pizzi dice la sua... L'estremo riciclaggio, a metà strada tra i «dischi per documen a convegno tarsi» e il revival «genuino* è ambientato appunto negli Anni Cin franco ."Montini; -Il cinema f»cUmM»li quanta, anzi nei «Fantastici Anni Cinquanta», come è stata battez della ItAI-TV. di Ugo Itui/o- PARIGI — Giudirandolo non zata la nuova collana della Fonit II sottotitolo è: «Dai fiori ai ROMA — 'Ciurma italiano lan;«Lospario della critica nei -accettabile», ma senza conte cantautori», cioè dal primo festival di San Remo (trasmesso per KII-X2»: questo il tema tiri con mass-media» di Morando ?Ho- starne il .carattere estetico-, radio) ai vari Modugno, Giacomo Rondinella, Gianni Mcccia, al una società pubblicitaria di vegno rhr si inaugurerà a Lec randini e Pietro Pintus; -L'edi Ora tornano celebrato Fred Buscaglione. -

Who's Who Legal: Thought Leaders
Who’s Who Legal: Thought Leaders - Global Elite 2020 Arbitration .................................................................................................................................... 4 Asset Recovery ............................................................................................................................ 5 Aviation - Contentious ................................................................................................................. 7 Aviation - Finance ........................................................................................................................ 7 Aviation - Regulatory ................................................................................................................... 8 Banking - Finance ........................................................................................................................ 9 Banking - Fintech ....................................................................................................................... 10 Banking - Regulatory ................................................................................................................. 10 Business Crime Defence - Corporates ...................................................................................... 11 Business Crime Defence - Individuals ....................................................................................... 12 Capital Markets - Debt and Equity ............................................................................................ -

War Brewing Over Listings
REGISTER ONLINE TODAY! AT WWW.NEMORTGAGEEXPO.COM Established 1872 WEEK OF MONDAY, OCTOBER 21, 2013 www.bankerandtradesman.com THE FINANCIAL SERVICES AND REAL ESTATE WEEKLY FOR MASSACHUSETTS A Publication of The Warren Group FADING ALLIANCES War Brewing COMMERCIAL INTERESTS Over Listings Cracks Forming In Real Estate A TALE OF TWO CITIES World Could Change Business Boston May Be Booming, But the Motor City’s Got Upside Too BY COLLEEN M. SULLIVAN BANKER & TRADESMAN STAFF WRITER BY SCOTT VAN VOORHIS downtown Detroit is on the move right now, even by rewing tensions between some of BANKER & TRADESMAN COLUMNIST Boston standards. the country’s biggest brokers and t might have helped had Bos- In fact, along with a pretty tough baseball team, this Bthe nation’s 900-odd multiple list- ton Mayor Thomas M. Me- scrappy underdog of American cities has something ing services (MLSs) may create a schism Inino taken all of two minutes that is in short supply these days in the Hub – massive that could shake up the entire real estate to Google “downtown Detroit” upside potential. industry. before trashing the Motor City. “The downtown Detroit market is as hot as I have As real estate portals like Zillow and In fact, instead of recom- ever seen it,” said downtown Detroit market expert Trulia have become more and more pop- mending Detroit be blown up David MacDonald, an executive vice president at SCOTT VAN VOORHIS ular with consumers (and Wall Street in- to start all over again, Bos- Jones Lang LaSalle. vestors), the rest of the real estate indus- ton’s long-time mayor might have even learned a Menino, of course, is not alone in taking pot try is scrambling to think up new ways thing or two about how low business costs are shots at Detroit. -

2011–2012 Honor Roll of Donors 1 Honor Roll of Donors Honor Roll of Donors Dear Alumni, Parents and Friends
HONOR ROLL OF DONORS SAINT ANSELM 2011–2012 Honor Roll of Donors 1 HONOR ROLL OF DONORS HONOR ROLL OF DONORS DEAR ALUMNI, PARENTS AND FRIENDS Forty-eight years ago, I first set foot on the campus of Saint Anselm College as a nervous and excited freshman. Yes, the campus looked very different than it does today, but the hallmarks of this great school are unchanged: the warm, Benedictine hospitality that greeted my family and me, and the commitment to distinctive, quality, Catholic education. For nearly 25 years, I have served as President of Saint Anselm College. I have watched the college grow, both in population and in physical size, as we added new buildings and programs. Each and every day, I look upon the campus with great pride—pride in our students who are learning to think critically and ethically in their chosen fields of study, and in our faculty who continue to learn so that young Anselmians have the latest information and the tools needed to succeed. I have pride in our staff members who go out of their way to help our students and graduates, and in our alumni themselves—more than 20,000 strong—who never cease to amaze me with their incredible accomplishments. And I have pride in you for your commitment to Saint Anselm College. This Annual Honor Roll of Donors honors you, our loyal donors, for your support. This book boasts 6,811 names, and is something I look upon with an overwhelming sense of thankfulness. I am truly grateful to all who have supported Saint Anselm and to all who plan to do so in the future. -
Clara Zabitski Mrs
Clara Zabitski Mrs. Clara M. Zabitski, 76, Martins Ferry, died Thursday morning, May 4. in Martins Ferry Hospital. She was born July 3, 1895, in Poland. She was a member of St. Mary’s Roman Catholic Church, Martins Ferry, and was preceded in death by her husband, Charles, in 1939, and a son, John, in 1965. Surviving are a son, Joseph of Martins Ferry, with whom she made her home, three daughters, Mrs. Ann Rykowski of Martins Ferry, Mrs. Sophia McKay of St. Clairsville, Mrs. Mario (Stella) Veneri of RD, Powhatan, two brothers, and one sister in Poland, 10 grand children and four great grand children. Mass of Resurrection was held Monday at St. Mary’s Church, Martins Ferry, with Msgr. Joseph J. Kloss as celebrant. Interment was in Riverview Cemetery, Martins Ferry. Elizabeth Zabitski Elizabeth J. Zabitski, 67, 237 N. 7lh St., Martins Ferry, died Monday in East Ohio Regional Hospital at Martins Ferry. She was bom Aug. 25, 1925 in Martins Ferry, daughter of the late George and Bertha Corise Micker. She was a former beautician and a member of St. Mary Catholic Church, Martins Ferny. She was preceded in death by her husband, Joseph Zabitski; a brother, Rudy Micker; a sister, Martha Micker. Surviving are a son, Joseph Zabit ski of Martins Ferry; a brother, George Micker of Martins Ferry; three sisters, Mary Micker of St. Clairsville, Bertha Holland of Mor ristown and Mrs. Edward (Ann) Rec tor of Harrisville; two grandchildren. Friends will be received 2 to 4 and 7 to 9 p.m. today at Keller Funeral Home, Martins Ferry, where services will be held at 11 a.m. -

Fi«I AHÜCÜCO- Sixtus 11 Gives the Treasures of the Church to the Deacon St
Fi«i AHÜCÜCO- Sixtus 11 gives the treasures of the church to the deacon St. Lawrence ^detail) Frsi An^elico prohahly took Eugenius IV as his model for this fresco of Sixtus II, - .is lie took Nicholas V (at his command Iv executed these paintings) for the other papal iigure in the decoration of the chapel of Pope Nicholas in the Vatican. THE POPES THROUGH HISTORY edited hy RAYMOND H. SCHMANDT Loyola University, Chicago Volume 1 ;Eugenius IV .Pope of Christian Union by JOSEPH GILL, S. J. Professor of the Pontifical Oriented Institute, Rome LONDON .URNS & OATES împnmi potest: ALPHONSUS RABS, S.J. February 8, 196Î Nihil Obstat: EDWARD A. CBRNY, S.S. Censor Librorum Imprimatur: FRANCIS P. KEOUGH, D.D. Archbishop of Baltimore October 10, 1961 The nihil obstat and imprimatur are official declarations that a book or pamphlet is free of doctrinal and moral error. No implication is contained therein that those who have granted the nihil obstat and imprimatur agree with the opinions expressed. Copyright © 1961 by THE NEWMAN PRESS Library of Congress Catalog Card Number: 61-16572 Printed in the United States of America Introduction to the Series IHIS VOLUME initiates a new series, "The Popes Through History/* a series consisting of biographies of the most important popes who reigned in times of particular crisis for the Church, Too many of the really significant popes are unknown outside of scholarly circles. Others are scarcely or inaccurately understood even by professional historians. Still others have fared so badly at the hands of apologists or of critics of the Papacy as a Catholic institution that the common view dis- torts them beyond recognition. -

Optimization of High-Throughput Multiplexed Phenotyping of Extracellular Vesicles Performed in 96-Well Microtiter Plates
polymers Article Optimization of High-Throughput Multiplexed Phenotyping of Extracellular Vesicles Performed in 96-Well Microtiter Plates Malene Møller Jørgensen 1,2,* , Jenni Kathrine Sloth 1 and Rikke Bæk 1 1 Department of Clinical Immunology, Aalborg University Hospital, 9000 Aalborg, Denmark; [email protected] (J.K.S.); [email protected] (R.B.) 2 Department of Clinical Medicine, Aalborg University, 9000 Aalborg, Denmark * Correspondence: [email protected] Abstract: Extracellular vesicles (EVs) are promising biomarkers for several diseases, however, no simple and robust methods exist to characterize EVs in a clinical setting. The EV Array analysis is based on a protein microarray platform, where antibodies are printed onto a solid surface that enables the capture of small EVs (sEVs) by their surface or surface-associated proteins. The EV Array analysis was transferred to an easily handled microtiter plate (MTP) format and a range of optimization experiments were performed within this study. The optimization was performed in a comprehensive analytical setup where the focus was on the selection of additives added to spotting-, blocking-, and incubation buffers as well as the storage of printed antibody arrays under different temperatures from one day to 12 weeks. After ending the analysis, the stability of the fluorescent signal was investigated at different storage conditions for up to eight weeks. The various parameters and conditions tested within this study were shown to have a high influence on each other. The reactivity of the spots was found to be preserved for up to 12 weeks when stored at room temperature and using blocking procedure IV in combination with trehalose in the spotting buffer. -

Bridgewater Magazine, Volume 7, Number 1, Fall 1996 Bridgewater State College
Bridgewater State University Virtual Commons - Bridgewater State University Bridgewater Magazine Campus Journals and Publications 1996 Bridgewater Magazine, Volume 7, Number 1, Fall 1996 Bridgewater State College Recommended Citation Bridgewater State College (1996). Bridgewater Magazine, Vol. 7, No. 1. Retrieved from http://vc.bridgew.edu/br_mag/42 This item is available as part of Virtual Commons, the open-access institutional repository of Bridgewater State University, Bridgewater, Massachusetts. .' Fall, 1996 Volume 7, No. 1 A Publication for Alumni, Parents, and Friends of Bridgewater State College Lou Ricciardi, '81 and Cynthia (Booth) Ricciardi, '81, at Alumni Park. He is the Chairman of the Bridgewater Foundation, she is President-Elect of the Alumni Association The Bridgewater State College Foundation Proudly Announces F E AL The Boys Choir of Harlem Saturday, December 14, 1996·8:00 PM Founded in 1968 in the basement of the Ephesus Church, The Bo,s Choir of Harlem has become an international phenomenon. Billboard Magazine says the "classically rich...voices take flight on a contemporary slice of pop!hip-hop." New York Newsda, says they have "become one of the definitive cultural landmarks in a city top-loaded with them." The Bo,s Choir of Harlem has Neil Sedaka appeared on 60 Minutes, CNN, The David Letterman Show, Saturday, March 22,1997·8:00 PM and on the sound tracks ofSpike Lee's "Jungle Fever", Few musicians have attained the commeocial "Malcolm-X", and the Grammy-winning "Glory." For a success awarded to Neil Sedaka. Pop hits like night of inspiration and angelic uplifting, don't miss the Oh Carol, Calendar Girl, and Love Will Keep "best known boys choir in the country." Us Together have sold millions of records and Orchestra Seats $29 • Balcony Seats $2S have earned him an international reputation PUCCiNI'S as a singer-songwriter and pop-pioneer. -
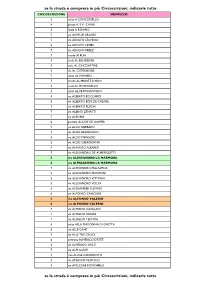
Se La Strada È Compresa in Più Circoscrizioni, Indicarle Tutte Se La
se la strada è compresa in più Circoscrizioni, indicarle tutte CIRCOSCRIZIONE INDIRIZZO 3 salita A CONCONELLO 4 piazza A. E K. CASALI 3 largo A ROIANO 7 via ACHILLE GRANDI 5 via ADOLFO LEGHISSA 6 via ADOLFO LEVIER 7 via ADOLFO REBEZ 7 vicolo AI PLAI 4 scala AL BELVEDERE 6 viale AL CACCIATORE 3 via AL CISTERNONE 7 salita AL MONBEU 7 strada AL MONTE D'ORO 3 scala AL MONTICELLO 4 salita AL PROMONTORIO 4 via ALBERTO BOCCARDI 6 via ALBERTO BOIS DE CHESNE 7 via ALBERTO PUSCHI 7 via ALBINO ZENATTI 7 via ALBONA 6 piazzale ALCIDE DE GASPERI 7 via ALDO BARBARO 7 via ALDO BRANDOLIN 4 via ALDO MANUZIO 3 via ALDO OBERDORFER 4 via ALEARDO ALEARDI 7 via ALESSANDRO DE ALMERIGOTTI 5 via ALESSANDRO LA MARMORA 6 via ALESSANDRO LA MARMORA 7 via ALESSANDRO MALASPINA 5 via ALESSANDRO MANZONI 5 via ALESSANDRO VITTORIA 3 via ALESSANDRO VOLTA 3 via ALEXANDER FLEMING 6 via ALFONSO CANCIANI 3 via ALFONSO VALERIO 6 via ALFONSO VALERIO 7 via ALFREDO CATALANI 5 via ALFREDO ORIANI 7 via ALFREDO TESTONI 3 salita ALLA MADONNA DI GRETTA 6 via ALLE CAVE 7 via ALLE TRE CROCI 6 androna ALMERICO D'ESTE 3 via ALMERIGO GRILZ 7 via ALPI GIULIE 7 riva ALVISE CADAMOSTO 5 via AMERIGO VESPUCCI 4 via AMILCARE PONCHIELLI se la strada è compresa in più Circoscrizioni, indicarle tutte se la strada è compresa in più Circoscrizioni, indicarle tutte CIRCOSCRIZIONE INDIRIZZO 7 via ANDREA ANTICO 5 via ANDREA MANTEGNA 5 via ANDREA PALLADIO 4 via ANDREA RAPICIO 7 via ANGELO CATTARUZZA 5 via ANGELO DE VALENTINI 6 via ANGELO DE VALENTINI 5 via ANGELO EMO 7 via ANGELO VIVANTE 2 via ANTON KUHELJ -

Ferulic Acid Derivatives and Avenanthramides Modulate Endothelial Function Through Maintenance of Nitric Oxide Balance in HUVEC Cells
nutrients Article Ferulic Acid Derivatives and Avenanthramides Modulate Endothelial Function through Maintenance of Nitric Oxide Balance in HUVEC Cells Gabriele Serreli 1,2,† , Melanie Le Sayec 1,†, Estelle Thou 1, Camille Lacour 1, Camilla Diotallevi 1, Misbah Arshad Dhunna 1, Monica Deiana 2 , Jeremy P. E. Spencer 3 and Giulia Corona 1,* 1 Health Sciences Research Centre, Life Sciences Department, Whitelands College, University of Roehampton, London SW15 4JD, UK; [email protected] (G.S.); [email protected] (M.L.S.); [email protected] (E.T.); [email protected] (C.L.); [email protected] (C.D.); [email protected] (M.A.D.) 2 Department of Biomedical Sciences, University of Cagliari, Cittadella Universitaria, SS 554, km 4.5, 09042 Monserrato, Italy; [email protected] 3 Molecular Nutrition Group, Food and Nutritional Sciences Department, University of Reading, Reading RG6 6AP, UK; [email protected] * Correspondence: [email protected] † These authors contributed equally to this work. Abstract: Wholegrain oats contain a variety of phenolic compounds thought to help maintain healthy vascular function, through the maintenance of local levels of the vasodilator nitric oxide (NO). Thus, the full molecular mechanisms involved are not yet clear. With this work we aim to understand Citation: Serreli, G.; Le Sayec, M.; the possible cellular mechanisms by which avenanthramides and ferulic acid derivatives, present Thou, E.; Lacour, C.; Diotallevi, C.; in oats, may help maintain a healthy vascular function through the modulation of the NO pathway. Dhunna, M.A.; Deiana, M.; Spencer, Primary Human Umbilical Vein Endothelial Cells (HUVEC) were exposed to ferulic acid, isoferulic J.P.E.; Corona, G. -

Bulletin Municipale FINALE.Pdf
Saint-Prim Bulletin municipal 2010 Liste des Rues Acacias (chemin des) C3 C4 Auberives (route d’) C4 D4 Bambous (allée des) B2 B3 Bellevue (rue de) A2 A3 Berbin (chemin de) C1 Buffon (rue de) B4 Carrosses (chemin des) A2 Cerisiers (chemin des) C3 D3 Chanet (rue du) C2 Châtaignier (chemin du) B1 C2 Corneyzin (chemin de) C1 Crazes (allée des) B2 C2 Crètes (rue des) A2 Croix Rouge (chemin de la) A2 Cyprès (allée des) B2 C2 Erables (allée des) B2 C2 Fontaine (chemin de la) D2 C2 Fontenettes (chemin des) D1 Glay (route de) B4 C3 Grandes Bruyères (ch. ) A2 B1 Lemps (chemin des) D3 Marquis (chemin du) B4 B3 Mordant (chemin de) A3 B3 Mûriers (allée des) D1 E1 Parc (allée du) B3 Pierres (chemin des) B2 Pré Margot (rue de) A1 A2 Providence (impasse de la) B3 Rivière (chemin de la) C4 Roches (route des) A2 B2 Rôtisses (chemin des) B2 Roussières (chemin des) A2 A3 Saint Clair (route de) A3 B2 Saint Jean (chemin de) D1 Saluant (chemin de) E2 Source (allée de la) B3 Vallon (chemin du) B2 Valottes (chemin des) B2 Vergers (chemin des) B4 Vienne (route de) C2 E2 Village (rue du) B2 Vignes (chemin des) A2 A3 SOMMAIRE Page Le mot du maire 4 Info pratique – Avis et recommandations 5 à 9 Etat civil 10 & 11 Budget et travaux 12 à 15 Chantier école (en photos) 16 Ecole 17 Restaurant scolaire et garderie 18 Relais assistantes maternelles 19 Sou des écoles 20 Patrimoine & « de croix en fontaines » 21 à 23 Le Mas des Champs 24 & 25 C.C.A.S. -

Town of Scarborough, ME
Town of Scarborough, ME Location Listing St# Street Name Map Lot Owner Name Use Code Use Description Old Value Proposed Value 5 ABBI LN R021 002E COOK, ANTHONY JOSEPH III 1010 SINGLE FAMILY 357,600 462,600 6 ABBI LN R021 002 CUNNINGHAM, JAMES 1010 SINGLE FAMILY 373,100 451,100 7 ABBI LN R021 002F KERKELA, LEIF A 1010 SINGLE FAMILY 354,700 446,300 8 ABBI LN R021 002N JOHNSON, LYNDA S 1300 VACANT LAND 84,200 123,000 9 ABBI LN R021 002M VECCHIONE, MICHAEL 1010 SINGLE FAMILY 328,600 461,200 10 ABBI LN R021 002I NEWCOMB, MATTHEW J 1010 SINGLE FAMILY 348,200 447,600 11 ABBI LN R021 002L DOBBINS, SARAH ELIZABETH 1010 SINGLE FAMILY 335,300 471,400 12 ABBI LN R021 002H STEPHENSON, ERIC M 1010 SINGLE FAMILY 359,800 472,900 14 ABBI LN R021 002G CAMP, MARK 1010 SINGLE FAMILY 374,200 513,600 16 ABBI LN R021 002J PAYEUR, NICHOLAS DAVID 1010 SINGLE FAMILY 374,300 475,900 18 ABBI LN R021 002K JOHNSON, MICHAEL E 1010 SINGLE FAMILY 399,300 576,600 1 ABIGAIL WAY U049 001 ALLEN, GREGG N 1010 SINGLE FAMILY 263,400 317,300 2 ABIGAIL WAY U048 017B WALLACE, LESTER A 1010 SINGLE FAMILY 422,400 521,100 3 ABIGAIL WAY R058 2601 MURPHY, PATRICIA A 1010 SINGLE FAMILY 450,700 482,300 5 ABIGAIL WAY R058 2602 CHAMBERS, TERRY L 1010 SINGLE FAMILY 460,300 432,800 6 ABIGAIL WAY R058 2619 MASON, SCOTTE & PAUL TRUST 1010 SINGLE FAMILY 422,700 460,800 7 ABIGAIL WAY R058 2603 DOHERTY, AMANDA J 1010 SINGLE FAMILY 408,700 447,300 8 ABIGAIL WAY R058 2618 CONNOLLY, JEFFREY J 1010 SINGLE FAMILY 399,000 455,900 9 ABIGAIL WAY R058 2604 LEE, STANLEY S 1010 SINGLE FAMILY 346,700 434,400