A New Route to Sugar Alcohols
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

GRAS Notice 789 for Erythritol
GRAS Notice (GRN) No. 789 https://www.fda.gov/food/generally-recognized-safe-gras/gras-notice-inventory. Toi• Strategies ~~~~G~~[)) JUN 7 20'8 Innovative solutions Sound science OFFICE OF FOOD ADDITIVE SAFE1Y June 5, 2018 Dr. Dennis Keefe Director, Division of Biotechnology and GRAS Notice Review Office of Food Additive Safety (HFS-200) Center for Food Safety and Applied Nutrition Food and Drug Administration 5100 Paint Branch Parkway College Park, MD 20740-3835 Subject: GRAS Notification - Erythritol Dear Dr. Keefe: On behalf of Cargill, Incorporated, ToxStrategies, Inc. (its agent) is submitting, for FDA review, a copy of the GRAS notification as required. The enclosed document provides notice of a claim that the food ingredient, erythritol, described in the enclosed notification is exempt from the premarket approval requirement of the Federal Food, Drug, and Cosmetic Act because it has been determined to be generally recognized as safe (GRAS), based on scientific procedures, for addition to food. If you have any questions or require additional information, please do not hesitate to contact me at 630-352-0303, or [email protected]. Sincerely, (b) (6) Donald F. Schmitt, M.P.H. Senior Managing Scientist ToxStrategies, Inc., 931 W. 75th St. , Suite 137, PMB 263, Naperville, IL 60565 1 Office (630) 352-0303 • www.toxstrategies.com GRAS Determination of Erythritol for Use in Human Food JUNES,2018 Innovative solutions s ,..,.,',--.r-.r--.r--. OFFICE OF FOOD ADDITIVE SAFE1Y GRAS Determination of Erythritol for Use in Human Food SUBMITTED BY: Cargill, Incorporated 15407 McGinty Road West Wayzata, MN 55391 SUBMITTED TO: U.S. Food and Drug Administration Center for Food Safety and Applied Nutrition Office of Food Additive Safety HFS-200 5100 Paint Branch Parkway College Park MD 20740-3835 CONTACT FOR TECHNICAL OR OTIIER INFORMATION Donald F. -
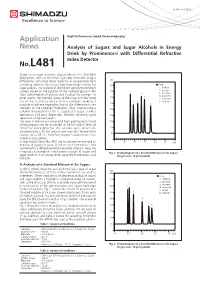
Analysis of Sugars and Sugar Alcohols in Energy Drink by Prominence-I with Differential Refreactive Index Detector
LAAN-A-LC-E258 Application High Performance Liquid Chromatography News Analysis of Sugars and Sugar Alcohols in Energy Drink by Prominence-i with Differential Refractive Index Detector No.L481 Sugars and sugar alcohols display almost no ultraviolet absorption, and are therefore typically detected using a differential refractive index detector or evaporative light uRI scattering detector. By using a ligand exchange column for 80 ■ Peaks sugar analysis, it is possible to distinguish among the different 1. maltose 70 2. glucose isomers based on the position of the hydroxyl group in the 1 4 3. fructose chair conformation of glucose and fructose for example. In 4. erythritol 60 5 other words, the hydroxyl group of the sugar and the metal 5. mannitol ion of the stationary phase form a complex, making it 2 3 6. sorbitol possible to achieve separation due to the difference in the 50 6 strength of the complex formation. Also, maintaining a 40 column temperature of 80 °C suppresses sugar anomer separation and peak dispersion, thereby achieving good 30 separation of adjacent peaks. The new Prominence-i integrated high-performance liquid 20 chromatograph can be connected to the RID-20A differential refractive index detector. The column oven, which can 10 accommodate a 30 cm column and maintain temperature 0 control up to 85 °C, therefore supports applications that require a long column. In Application News No. 467, we introduced an example of 0 5 10 15 20 25 min analysis of sugars in juice, in which the Prominence-i was connected to a differential refractive index detector. Here, we introduce an example of simultaneous analysis of sugars and Fig. -

United States Patent Office
United States Patentaw Office ... ber of ether groupings on each molecule. These mix tures, however, if they analyze as containing an average 2,978,421. number of ether groups per molecule greater than one, NITRILE COPOLYMERS AND METHOD OF are capable of producing the insoluble nitrile polymers PREPARING SAME of this invention. Since the efficiency of the polyether cross-linking agent increases with the number of poten John A. Holloway, Cleveland, Ohio, assignor to The tially polymerizable groups on the molecule, it is much B. F. Goodrich Company, New York, N.Y., a corpo preferred to utilize polyethers containing an average of ration of New York - two or more alkenyl ether groupings per molecule. The No Drawing. Filed June 9, 1958, Ser. No. 740,566. 10 polyvinyl polyethers of the polyhydric alcohols within the above broad class are produced by reacting acetylene 9 Claims. (C. 260-17.4) with the polyhydric alcohol (or an alcoholate thereof) in a Reppe-type vinylation synthesis. The polycrotyl ethers of the polyhydric alcohols are also useful although they This invention relates to cross-linked alpha-beta ole 15 do not contain a terminal CHCC group. - finically unsaturated nitrile polymers and more particu Illustrative polyhydric alcohols of the above-described larly pertains to copolymers of alpha-beta olefinically class that may be utilized in the preparation of the poly. unsaturated nitriles and polyalkenyl polyethers of poly alkenyl polyether, cross-linking agent include the butane hydric alcohols and methods for their preparation. triols such as 1,2,3-butane triol, 2,3,4-trihydroxybutyric An object of this invention is the provision of insoluble, 20 acid, the aldotetroses such as erythrose and threose, keto cross-linked polynitriles which are capable of thickening tetroses such as erythrulose; the aldopentoses such as certain non-aqueous polar solvents. -

LIQUID-VAPOR EQUILIBRIUM THERMODYNAMICS of the FUSEL OIL: a CASE STUDY Jéssyka Jennifer Miranda Corrêa¹, Edilailsa Januário
LIQUID-VAPOR EQUILIBRIUM THERMODYNAMICS OF THE FUSEL OIL: A CASE STUDY Jéssyka Jennifer Miranda Corrêa¹, Edilailsa Januário de Melo², José Izaquiel Santos da Silva³ ¹Graduating in Chemical Engineering from the Federal University of the Jequitinhonha and Mucuri Valleys, Diamantina - MG, Brazil. ²PhD student in Chemical Engineering, College of Chemical Engineering - State University of Campinas - SP, Brazill. ³Professor at the Federal University of the Jequitinhonha and Mucuri Valleys, Diamantina - MG, Brazil. E-mail for contact: [email protected] Recebido em: 06/04/2019 – Aprovado em: 10/06/2019 – Publicado em: 30/06/2019 DOI: 10.18677/EnciBio_2019A183 ABSTRACT Fusel oil is a by-product of the process of recovering hydrated ethanol, consisting of a mixture of superior alcohols, ethanol, water, among other components. Its commercial interest is mainly due to the presence of isoamyl alcohol, one of the raw materials used in the synthesis of esters, which compounds are relevant for the chemical industry. The recovery of this superior alcohol involves unit operations based on phase equilibrium, which for multicomponent systems can be studied in software that simulates the operation of equipment, in order to present predictions of interactions between its components, reducing errors, time and costs of design. In the industrial, academic and scientific fields, there are numerous challenges regarding the design of separation processes and the monitoring of mixing effects in typical operations, and the Aspen Plus process simulation and optimization platform excels in solving these issues. The current study aims to predict the Liquid-Vapor Equilibrium of the fusel oil, which was considered as a mixture of five components, from the behavioral analysis of each of the binaries that compose it. -

IPOLYHYDRIC ALCOHOLS from WOOD July 1954
IPOLYHYDRIC ALCOHOLS FROM WOOD July 1954 DEC 221954 No. 1984 UNITED STATES DEPARTMENT OF AGRICULTURE --,LFOREST. SERVICE 40REST PRODUCTS LABORATORY÷Q-.---,Y) Madison 5,Wisconsin In Cooperation with the University of Wisconsin 1 POLYHYDRIC ALCOHOLS FROM WOOD-2 2— By J. A. HALL, Director Forest Products Laboratory, 3 Forest Service U. S. Department of Agriculture Wood-sugar solutions produced in the laboratory-scale pilot plant at the U. S. Forest Products Laboratory have been proven to be satis- factory for the production of industrial alcohol, feeding yeast, and wood 4 molasses for stock feed (14, 15, 16, P 17). ± As a continuation of the pro- gram of the Laboratory for developing chemical utilization processes for the wood residues resulting from normal logging operations, and lumber, veneer, and the other forest products manufacture, investiga- tions are in progress to convert wood sugar into merchantable products. Because of the versatility and greatly increased demand for polytlydric alcohols, a study of the production of these materials has been chosen. The alkyd resin industry in the United States has increased tenfold in the past few years and no evidence of decline has been noted. Recently sorbitol has been made available commercially by the catalytic hydro- genation of corn sugar (2). The annual capacity now is 75 million pounds. Sorbitol is widely used as a humectant and conditioner in tobacco, cello- phane, and other products (3). However, one of its fast growing uses is for alkyd resins, where it may replace up to 50 percent glycerine and 25 percent pentaerythritol. This is fortunate since the production of glycerine in the United States as a byproduct of the soap manufacturing 1Presented before the Food and Agriculture Organization of the United Nations, Expert Panel on the Chemistry of Wood, meeting in Stockholm, Sweden, July 27-28, 1953. -

Sugar Alcohols—Their Role in the Modern World of Sweeteners: a Review
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Springer - Publisher Connector Eur Food Res Technol (2015) 241:1–14 DOI 10.1007/s00217-015-2437-7 REVIEW PAPER Sugar alcohols—their role in the modern world of sweeteners: a review Małgorzata Grembecka Received: 31 August 2014 / Revised: 16 December 2014 / Accepted: 13 February 2015 / Published online: 28 February 2015 © The Author(s) 2015. This article is published with open access at Springerlink.com Abstract Epidemic obesity and diabetes encouraged the observed. It was mainly due to the developments in bio- changes in population lifestyle and consumers’ food prod- logical studies, the change of a population lifestyle and the ucts awareness. Food industry has responded people’s increase in the consumer awareness concerning food prod- demand by producing a number of energy-reduced prod- ucts. The health quality of food depends mainly on nutri- ucts with sugar alcohols as sweeteners. These compounds ents, but also on foreign substances such as food additives. are usually produced by a catalytic hydrogenation of carbo- The presence of foreign substances in the food can be justi- hydrates, but they can be also found in nature in fruits, veg- fied, allowed or tolerated only when they are harmless to etables or mushrooms as well as in human organism. Due our health. Epidemic obesity and diabetes encouraged the to their properties, sugar alcohols are widely used in food, growth of the artificial sweetener industry. There are more beverage, confectionery and pharmaceutical industries and more people who are trying to lose weight or keeping throughout the world. -

Sugar Alcohols: a Review
International Journal of PharmTech Research CODEN (USA): IJPRIF, ISSN: 0974-4304, ISSN(Online): 2455-9563 Vol.9, No.7, pp 407-413, 2016 Sugar alcohols: A review Ramezan Ali Mahian1, Vahid Hakimzadeh2* 1,2Department of Food Science and Technology, Quchan Branch, Islamic Azad University, Quchan, Iran Abstract : Sugar alcohols are extensively used as sweetening agents. They sometimes possess advantages over the parent sugars in sweetness, caloric reduction and non-cariogenicity. The physical status of carbohydrates in food and confectionery affects both the properties of the product during production and the quality of the final product. Sugar alcohols, such as xylitol, mannitol, sorbitol, and erythritol are emerging food ingredients that provide similar or better sweetness/sensory properties of sucrose, but are less calorigenic. Also, sugar alcohols can be converted into commodity chemicals through chemical catalysis. Biotechnological production offers the safe and sustainable supply of sugar alcohols from renewable biomass. These compounds are usually produced by a catalytic hydrogenation of carbohydrates, but they can be also found in nature in fruits, vegetables or mushrooms as well as in human organism. Due to their properties, sugar alcohols are widely used in food, beverage, confectionery and pharmaceutical industries throughout the world. They have found use as bulk sweeteners that promote dental health and exert prebiotic effect. They are added to foods as alternative sweeteners what might be helpful in the control of calories intake. Consumption of low-calorie foods by the worldwide population has dramatically increased, as well as health concerns associated with the consequent high intake of sweeteners. Key words: Sugar alcohols, low-calorie, Sugar-free products, sweetener. -

(12) Patent Application Publication (10) Pub. No.: US 2015/0017246 A1 Huang (43) Pub
US 2015.0017246A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2015/0017246 A1 Huang (43) Pub. Date: Jan. 15, 2015 (54) NOVEL CROSSLINKING REAGENTS, Publication Classification MACROMOLECULES, THERAPEUTIC BIOCONJUGATES, AND SYNTHETIC (51) Int. Cl. METHODS THEREOF A647/48 (2006.01) CI2N II/06 (2006.01) (71) Applicant: CellMosaic, Inc., Worcester, MA (US) (52) U.S. Cl. CPC ............ A61K 47/4823 (2013.01): CI2N II/06 (72) Inventor: Yumei Huang, Lexington, MA (US) (2013.01); A61K 47/48369 (2013.01); A61 K 47/48238 (2013.01) USPC ....... 424/490; 536/1.11: 435/181: 530/391.9; (73) Assignee: CellMosaic, Inc., Worcester, MA (US) 530/402:536/6.4 (57) ABSTRACT (21) Appl. No.: 14/156.224 The invention provides novel chemical entities based on Sugar alcohols. These new chemical entities are biocompat ible and biodegradable. The molecules can be made in a (22) Filed: Jan. 15, 2014 single and pure form. The molecular weights of these mol ecules range from small (<1000 Da) to large (1000-120,000 Da). The Sugar alcohol-based molecules can have functional Related U.S. Application Data groups throughout the molecule for crosslinking compounds, Continuation-in-part of application No. PCT/US2012/ Such as the preparation of antibody-drug conjugates, or to (63) facilitate the delivery of therapeutic proteins, peptides, 047255, filed on Jul. 18, 2012. siRNA, and chemotherapeutic drugs. Also provided are new (60) Provisional application No. 61/509,296, filed on Jul. conjugate entities prepared through Sugar alcohol molecules. 19, 2011, provisional application No. 61/754,571, Methods of synthesizing Sugar alcohol-based molecules and filed on Jan. -
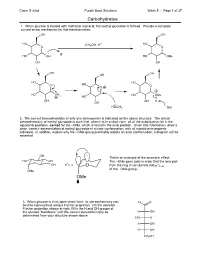
5.01 Carbohydrates Answers.Cdx
Chem S-20ab Purple Book Solutions Week 5 - Page 1 of 37 Carbohydrates 1. When glucose is treated with methanol and acid, the methyl glucoside is formed. Provide a complete curved-arrow mechanism for this transformation: OH OH HO CH OH, H+ HO O 3 O H+ HO OH HO OMe OH OH OH OH OH HO HO O HO O O HO OH HO OMe 2 HO OH OH H OH HOCH3 Sol 2. The correct stereochemistry of only one stereocenter is indicated on the above structure. The actual stereochemistry of methyl glucoside is such that, when it is in a chair form, all of the substituents fall in the equatorial positions, except for the –OMe, which is found in the axial position. Given this information, draw a clear, correct representation of methyl glucoside in a chair conformation, with all substituents properly indicated. In addition, explain why the –OMe group preferably adopts an axial conformation; a diagram will be essential. OH This is an example of the anomeric effect. HO OH The –OMe goes axial in order that the lone pair O * OH C–O O fromtheringOcandonateinto *C–O of the –OMe group. OMe OMe 3. When glucose is in its open-chain form, its stereochemistry can H O best be represented using a Fischer projection. On the skeleton Fischer projection shown at right, fill in the H and OH groups of the glucose "backbone" with the correct stereochemistry as H OH determined from your structure shown above. HO H H OH H OH CH2OH Chem S-20ab Purple Book Solutions Week 5 - Page 2 of 37 Carbohydrates: Mechanisms 1. -

Gut Microbiota Prevents Sugar Alcohol-Induced Diarrhea
nutrients Article Gut Microbiota Prevents Sugar Alcohol-Induced Diarrhea Kouya Hattori 1,2 , Masahiro Akiyama 1, Natsumi Seki 1,2, Kyosuke Yakabe 1,2, Koji Hase 2 and Yun-Gi Kim 1,* 1 Research Center for Drug Discovery, Faculty of Pharmacy and Graduate School of Pharmaceutical Sciences, Keio University, Tokyo 105-8512, Japan; [email protected] (K.H.); [email protected] (M.A.); [email protected] (N.S.); [email protected] (K.Y.) 2 Department of Biochemistry, Faculty of Pharmacy and Graduate School of Pharmaceutical Sciences, Keio University, Tokyo 105-8512, Japan; [email protected] * Correspondence: [email protected] Abstract: While poorly-absorbed sugar alcohols such as sorbitol are widely used as sweeteners, they may induce diarrhea in some individuals. However, the factors which determine an individual’s susceptibility to sugar alcohol-induced diarrhea remain unknown. Here, we show that specific gut bacteria are involved in the suppression of sorbitol-induced diarrhea. Based on 16S rDNA analysis, the abundance of Enterobacteriaceae bacteria increased in response to sorbitol consumption. We found that Escherichia coli of the family Enterobacteriaceae degraded sorbitol and suppressed sorbitol-induced diarrhea. Finally, we showed that the metabolism of sorbitol by the E. coli sugar phosphotransferase system helped suppress sorbitol-induced diarrhea. Therefore, gut microbiota prevented sugar alcohol-induced diarrhea by degrading sorbitol in the gut. The identification of the gut bacteria which respond to and degrade sugar alcohols in the intestine has implications for microbiome science, processed food science, and public health. Keywords: gut microbiota; sugar alcohol; diarrhea Citation: Hattori, K.; Akiyama, M.; Seki, N.; Yakabe, K.; Hase, K.; Kim, Y.-G. -

Sugar Alcohols Fact Sheet
International Food Information Council Sugar Alcohols Fact Sheet September 2004 BACKGROUND Sugar alcohols or polyols, as they are also called, have a long history of use in a wide variety of foods. Recent technical advances have added to the range of sugar alcohols available for food use and expanded the applications of these sugar replacers in diet and health-oriented foods. They have been found useful in sugar-free and reduced-sugar products, in foods intended for individuals with diabetes, and most recently in new products developed for carbohydrate controlled eating plans. Sugar alcohols are neither sugars nor alcohols. They are carbohydrates with a chemical structure that partially resembles sugar and partially resembles alcohol, but they don’t contain ethanol as alcoholic beverages do. They are incompletely absorbed and metabolized by the body, and consequently contribute fewer calories. The polyols commonly used include sorbitol, mannitol, xylitol, maltitol, maltitol syrup, lactitol, erythritol, isomalt and hydrogenated starch hydrolysates. Their calorie content ranges from 1.5 to 3 calories per gram compared to 4 calories per gram for sucrose or other sugars. Most are approximately half as sweet as sucrose; maltitol and xylitol are about as sweet as sucrose. Sugar alcohols occur naturally in a wide variety of fruits and vegetables, but are commercially produced from other carbohydrates such as sucrose, glucose, and starch. Along with adding a sweet taste, polyols perform a variety of functions such as adding bulk and texture, providing a cooling effect or taste, inhibiting the browning that occurs during heating and retaining moisture in foods. While polyols do not actually prevent browning, they do not cause browning either. -

Sugar Alcohols and Dental Caries
Federal Register / Vol. 61, No. 165 / Friday, August 23, 1996 / Rules and Regulations 43433 4. Paragraph (b) of § 210.76 is revised sugar alcohols to the nonpromotion of evaluate such claims, that the claim is to read as follows: dental caries are justified. FDA is supported by such evidence (see also announcing these actions in response to § 101.14(c)). FDA considered the § 210.76 Modification or rescission of a petition filed by the National relevant scientific studies and data exclusion orders, cease and desist orders, and consent orders. Association of Chewing Gum presented in the petition as part of its Manufacturers, Inc., and an ad hoc review of the scientific literature on * * * * * working group of sugar alcohol sugar alcohols and dental caries. The (b) Commission action upon receipt of manufacturers (hereinafter referred to as agency summarized this evidence in the petition. The Commission may the petitioners). proposed rule (60 FR 37507). thereafter institute a proceeding to DATES: Effective January 1, 1998. The The proposed rule included modify or rescind the exclusion order, qualifying and disqualifying criteria for cease and desist order, or consent order Director of the Office of the Federal Register approves the incorporation by the purpose of identifying foods eligible by issuing a notice. The Commission to bear a health claim. The proposal also may hold a public hearing and afford reference in accordance with 5 U.S.C. 552(a) and 1 CFR part 51 of a certain specified mandatory content and label interested persons the opportunity to information for health claims statements appear and be heard.