Genome Wide Analysis Across Alzheimer's Disease
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Datasheet Blank Template
SAN TA C RUZ BI OTEC HNOL OG Y, INC . Fe65L (Y-14): sc-104237 BACKGROUND PRODUCT Fe65L (Fe65-like protein), also known as APBB2 (amyloid β (A4) precursor Each vial contains 200 µg IgG in 1.0 ml of PBS with < 0.1% sodium azide protein-binding, family B, member 2), is a 758 amino acid protein that contains and 0.1% gelatin. one WW domain and 2 PID domains. Binding to the intracellular domain of Blocking peptide available for competition studies, sc-104237 P, (100 µg the -amyloid precursor protein, Fe65L is thought to modulate the internal - β peptide in 0.5 ml PBS containing < 0.1% sodium azide and 0.2% BSA). ization and, therefore, the accessibility and function of β-amyloid. Via its ability to control the intracellular accumulation of -amyloid, Fe65L is thought β APPLICATIONS to play a role in the pathogenesis of Alzheimer’s disease. Multiple isoforms of Fe65L exist due to alternative splicing events. The gene encoding Fe65L Fe65L (Y-14) is recommended for detection of Fe65L of mouse, rat and maps to human chromosome 4, which encodes nearly 6% of the human human origin by Western Blotting (starting dilution 1:200, dilution range genome and has the largest gene deserts (regions of the genome with no 1:100-1:1000), immunoprecipitation [1-2 µg per 100-500 µg of total protein protein encoding genes) of all of the human chromosomes. Defects in some (1 ml of cell lysate)], immunofluorescence (starting dilution 1:50, dilution of the genes located on chromosome 4 are associated with Huntington’s range 1:50-1:500) and solid phase ELISA (starting dilution 1:30, dilution dis ease, Ellis-van Creveld syndrome, methylmalonic acidemia and polycystic range 1:30-1:3000). -

Viewed and Published Immediately Upon Acceptance Cited in Pubmed and Archived on Pubmed Central Yours — You Keep the Copyright
BMC Genomics BioMed Central Research article Open Access Differential gene expression in ADAM10 and mutant ADAM10 transgenic mice Claudia Prinzen1, Dietrich Trümbach2, Wolfgang Wurst2, Kristina Endres1, Rolf Postina1 and Falk Fahrenholz*1 Address: 1Johannes Gutenberg-University, Institute of Biochemistry, Mainz, Johann-Joachim-Becherweg 30, 55128 Mainz, Germany and 2Helmholtz Zentrum München – German Research Center for Environmental Health, Institute for Developmental Genetics, Ingolstädter Landstraße 1, 85764 Neuherberg, Germany Email: Claudia Prinzen - [email protected]; Dietrich Trümbach - [email protected]; Wolfgang Wurst - [email protected]; Kristina Endres - [email protected]; Rolf Postina - [email protected]; Falk Fahrenholz* - [email protected] * Corresponding author Published: 5 February 2009 Received: 19 June 2008 Accepted: 5 February 2009 BMC Genomics 2009, 10:66 doi:10.1186/1471-2164-10-66 This article is available from: http://www.biomedcentral.com/1471-2164/10/66 © 2009 Prinzen et al; licensee BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Abstract Background: In a transgenic mouse model of Alzheimer disease (AD), cleavage of the amyloid precursor protein (APP) by the α-secretase ADAM10 prevented amyloid plaque formation, and alleviated cognitive deficits. Furthermore, ADAM10 overexpression increased the cortical synaptogenesis. These results suggest that upregulation of ADAM10 in the brain has beneficial effects on AD pathology. Results: To assess the influence of ADAM10 on the gene expression profile in the brain, we performed a microarray analysis using RNA isolated from brains of five months old mice overexpressing either the α-secretase ADAM10, or a dominant-negative mutant (dn) of this enzyme. -

Integrated Transcriptomic and Neuroimaging Brain Model Decodes
RESEARCH ARTICLE Integrated transcriptomic and neuroimaging brain model decodes biological mechanisms in aging and Alzheimer’s disease Quadri Adewale1,2,3, Ahmed F Khan1,2,3, Felix Carbonell4, Yasser Iturria-Medina1,2,3*, Alzheimer’s Disease Neuroimaging Initiative† 1Neurology and Neurosurgery Department, Montreal Neurological Institute, McGill University, Montreal, Canada; 2McConnell Brain Imaging Centre, Montreal Neurological Institute, McGill University, Montreal, Canada; 3Ludmer Centre for Neuroinformatics and Mental Health, McGill University, Montreal, Canada; *For correspondence: 4Biospective Inc, Montreal, Canada [email protected] †Data used in preparation of this article were partly obtained from the Alzheimer’s Disease Abstract Both healthy aging and Alzheimer’s disease (AD) are characterized by concurrent Neuroimaging Initiative (ADNI) alterations in several biological factors. However, generative brain models of aging and AD are database (adni.loni.usc.edu). As limited in incorporating the measures of these biological factors at different spatial resolutions. such, the investigators within the Here, we propose a personalized bottom-up spatiotemporal brain model that accounts for the ADNI contributed to the design direct interplay between hundreds of RNA transcripts and multiple macroscopic neuroimaging and implementation of ADNI modalities (PET, MRI). In normal elderly and AD participants, the model identifies top genes and/or provided data but did not modulating tau and amyloid-b burdens, vascular flow, glucose metabolism, functional activity, and participate in analysis or writing atrophy to drive cognitive decline. The results also revealed that AD and healthy aging share of this report. A complete listing specific biological mechanisms, even though AD is a separate entity with considerably more altered of ADNI investigators can be pathways. -
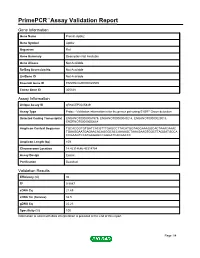
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name Protein Apbb2 Gene Symbol Apbb2 Organism Rat Gene Summary Description Not Available Gene Aliases Not Available RefSeq Accession No. Not Available UniGene ID Not Available Ensembl Gene ID ENSRNOG00000025509 Entrez Gene ID 305338 Assay Information Unique Assay ID qRnoCEP0025849 Assay Type Probe - Validation information is for the primer pair using SYBR® Green detection Detected Coding Transcript(s) ENSRNOT00000067979, ENSRNOT00000049214, ENSRNOT00000029018, ENSRNOT00000050664 Amplicon Context Sequence TGCACGGTGTGATCACGTTTGAGCCTTACATGGTAGGAAAGGCACTAAACAAAC TGAAGGAATGAGAACACAGGGCACCAAAAGCTAAAGAAGTGGCTTAGGATGCCA CCGAAGTCCATGAGAGCCGGGATCACGACCC Amplicon Length (bp) 109 Chromosome Location 14:43314646-43314784 Assay Design Exonic Purification Desalted Validation Results Efficiency (%) 94 R2 0.9987 cDNA Cq 21.69 cDNA Tm (Celsius) 82.5 gDNA Cq 25.23 Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 1/4 PrimePCR™Assay Validation Report Apbb2, Rat Amplification Plot Amplification of cDNA generated from 25 ng of universal reference RNA Melt Peak Melt curve analysis of above amplification Standard Curve Standard curve generated using 20 million copies of template diluted 10-fold to 20 copies Page 2/4 PrimePCR™Assay Validation Report Products used to generate validation data Real-Time PCR Instrument CFX384 Real-Time PCR Detection System Reverse Transcription Reagent iScript™ Advanced cDNA Synthesis Kit for RT-qPCR Real-Time PCR Supermix SsoAdvanced™ SYBR® Green Supermix Experimental Sample qPCR Reference Total RNA Data Interpretation Unique Assay ID This is a unique identifier that can be used to identify the assay in the literature and online. Detected Coding Transcript(s) This is a list of the Ensembl transcript ID(s) that this assay will detect. -

Mir‑205‑3P Promotes Lung Cancer Progression by Targeting APBB2
MOLECULAR MEDICINE REPORTS 24: 588, 2021 miR‑205‑3p promotes lung cancer progression by targeting APBB2 LING‑BIN XU1, JIE XIONG1, YA‑HUI ZHANG2, YUN DAI3, XIAO‑PING REN1, YA‑JUAN REN1, DONG HAN1, SHENG‑HONG WEI1 and MIN QI4 1Department of Pulmonary and Critical Care Medicine No. 2, Shaanxi Provincial People's Hospital, Xi'an, Shaanxi 710068; 2Department of Orthopaedics, Xi'an Daxing Hospital, Xi'an, Shaanxi 710016; 3Department of Thoracic Surgery and 4Imaging Center, Shaanxi Provincial People's Hospital, Xi'an, Shaanxi 710068, P.R. China Received August 26, 2020; Accepted March 29, 2021 DOI: 10.3892/mmr.2021.12227 Abstract. Non‑small cell lung cancer (NSCLC), a leading treatments, the 5‑year survival rate has remained as low as cause of cancer‑associated mortality, has resulted in low 15% since the 1970s (5). Therefore, further investigations into survival rates and a high mortality worldwide. Accumulating the mechanisms of NSCLC development and progression are evidence has suggested that microRNAs (miRs) play critical of the utmost importance for the diagnosis, prevention and roles in the regulation of cancer progression and the present treatment of this disease. study aimed to explore the underlying mechanism of miR‑205 MicroRNAs (miRNAs/miRs), a class of endogenous in NSCLC. Reverse transcription‑quantitative PCR was non‑coding RNAs, can mediate various gene expression levels performed, which determined that miR‑205 expression was by suppressing the translation or accelerating the decay of the upregulated in NSCLC, and the present study detected the target mRNA (6,7). Previous data have revealed that miRNAs upregulation of miR‑205‑3p in a number of NSCLC cell lines can regulate 30% of mRNAs (8). -

Non Commercial Use Only
Trends in Evolutionary Biology 2017; volume 6:6514 Potential impact tions including insertional mutagenesis, of primate-specific SVA generation of deletions at the insertion site, Correspondence: Olga Vasieva, Institute of 3’ or 5’ transduction events, non-allelic Integrative Biology, Department of retrotransposons during homologous recombination and Comparative Genomics, University of the evolution of human exonisation.1-4 More than 10,000 TE inser- Liverpool, Liverpool, L69 7ZB, UK. cognitive function tions occurred in the human genome since Tel.: +44.151.795.4456 - Fax: +44.151.795.4406. human-chimpanzee divergence which were E-mail: [email protected] suggested to have implications on human 1 1 Key words: SVA, retrotransposons, hominoid, Olga Vasieva, Sultan Cetiner, evolution and especially its reproductive, 2 3 brain, evolution. Abigail Savage, Gerald G. Schumann, cognitive and immune functions that Vivien J. Bubb,2 John P. Quinn2 diverged strongly during a relative short Contributions: OV conceived of the study, 1Institute of Integrative Biology, time period.4,5 The large number of such designed and performed the systems analysis, University of Liverpool, UK; TEs in the genome makes an analysis of and drafted the manuscript, SC performed the 2Department of Molecular and Clinical their specific contribution to evolution very computational analysis and took part in draft- ing of the manuscript, AS has performed the Pharmacology, Institute of Translational difficult, and previous studies highlighted retrieval of SVA-associated gene -

An Application for CEN Module Extraction And
www.nature.com/scientificreports OPEN THD-Module Extractor: An Application for CEN Module Extraction and Interesting Gene Received: 30 June 2016 Accepted: 03 November 2016 Identification for Alzheimer’s Published: 30 November 2016 Disease Tulika Kakati1, Hirak Kashyap2 & Dhruba K. Bhattacharyya1 There exist many tools and methods for construction of co-expression network from gene expression data and for extraction of densely connected gene modules. In this paper, a method is introduced to construct co-expression network and to extract co-expressed modules having high biological significance. The proposed method has been validated on several well known microarray datasets extracted from a diverse set of species, using statistical measures, such as p and q values. The modules obtained in these studies are found to be biologically significant based on Gene Ontology enrichment analysis, pathway analysis, and KEGG enrichment analysis. Further, the method was applied on an Alzheimer’s disease dataset and some interesting genes are found, which have high semantic similarity among them, but are not significantly correlated in terms of expression similarity. Some of these interesting genes, such as MAPT, CASP2, and PSEN2, are linked with important aspects of Alzheimer’s disease, such as dementia, increase cell death, and deposition of amyloid-beta proteins in Alzheimer’s disease brains. The biological pathways associated with Alzheimer’s disease, such as, Wnt signaling, Apoptosis, p53 signaling, and Notch signaling, incorporate these interesting genes. The proposed method is evaluated in regard to existing literature. The volume of data obtained by microarray or other new high throughput technologies have been increasing enormously, and as a result, the biological databases are growing enormously. -

Network-Based Identification of Genetic Factors in Ageing, Lifestyle
bioRxiv preprint doi: https://doi.org/10.1101/482844; this version posted November 29, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-ND 4.0 International license. Network-based identification of genetic factors in Ageing, lifestyle and Type 2 Diabetes that Influence in the progression of Alzheimer’s disease 1st Utpala Nanda Chowdhury 2nd Md. Babul Islam Department of Computer Science and Engineering Department of Applied Physics and Electronic Engineering University of Rajshahi University of Rajshahi Rajshahi, Bangladesh Rajshahi, Bangladesh [email protected] [email protected] 3rd Shamim Ahmad 5th Mohammad Ali Moni Department of Computer Science and Engineering Discipline of Biomedical Science, Faculty of Medicine and Health University of Rajshahi The University of Sydney Rajshahi, Bangladesh NSW, Australia shamim [email protected] [email protected] Abstract—Alzheimer’s disease (AD) is an incurable disease, I. INTRODUCTION and the causative risk factors, especially the modifiable ones, are still poorly understood which impedes the effective prevention Alzheimer’s disease (AD) is the most common form of and remedy strategies. We proposed a network-based quanti- tative framework to reveal the complex relationship of various dementia characterized by gradual degeneration in memory, biasing genetic factors for the AD. We analyzed gene expression thinking, language, and learning capacity [1]. Initial signs microarray data from the AD, ageing, severe alcohol consump- begin with the inability of forming recent memories, but tion, type II diabetes, high body fat, high dietary fat, obesity, di- inevitably affect all cognitive functions resulting in complete etary red meat, sedentary lifestyle, smoking, and control datasets. -

APBB2) Alternative Splicing
Biochimica et Biophysica Acta 1833 (2013) 1511–1526 Contents lists available at SciVerse ScienceDirect Biochimica et Biophysica Acta journal homepage: www.elsevier.com/locate/bbamcr A novel snRNA-like transcript affects amyloidogenesis and cell cycle progression through perturbation of Fe65L1 (APBB2) alternative splicing Ilaria Penna a, Irene Vassallo a, Mario Nizzari b, Debora Russo c, Delfina Costa a, Paola Menichini c, Alessandro Poggi c, Claudio Russo d, Giorgio Dieci e, Tullio Florio b,f, Ranieri Cancedda a,c, Aldo Pagano a,c,⁎ a Dept. of Experimental Medicine (DIMES), University of Genova, Genova, Italy b Dept. of Internal Medicine (DIMI), University of Genova, Genova, Italy c IRCCS-AOU San Martino-IST, Genova, Italy d Dept. of Health Sciences, University of Molise, Campobasso, Italy e Dept. of Molecular Biology and Biochemistry, University of Parma, Parma, Italy f Center of Excellence for Biomedical Research (CEBR), University of Genova, Italy article info abstract Article history: FE65 proteins constitute a family of adaptors which modulates the processing of amyloid precursor protein Received 13 August 2012 and the consequent amyloid β production. Thus, they have been involved in the complex and partially un- Received in revised form 17 January 2013 known cascade of reactions at the base of Alzheimer's disease etiology. However, FE65 and FE65-like proteins Accepted 18 February 2013 may be linked to neurodegeneration through the regulation of cell cycle in post-mitotic neurons. In this work Available online 26 February 2013 we disclose novel molecular mechanisms by which APBB2 can modulate APP processing. We show that APBB2 mRNA splicing, driven by the over-expression of a novel non-coding RNA named 45A, allow the generation of alter- Keywords: β APBB2 alternative splicing native protein forms endowed with differential effects on A production, cell cycle control, and DNA damage re- Cell cycle progression non-coding RNA sponse. -

Association of Genetic Variants with Primary Open-Angle Glaucoma Among Individuals with African Ancestry
UCLA UCLA Previously Published Works Title Association of Genetic Variants With Primary Open-Angle Glaucoma Among Individuals With African Ancestry. Permalink https://escholarship.org/uc/item/4102f35x Journal JAMA, 322(17) ISSN 0098-7484 Authors Genetics of Glaucoma in People of African Descent (GGLAD) Consortium Hauser, Michael A Allingham, R Rand et al. Publication Date 2019-11-01 DOI 10.1001/jama.2019.16161 Peer reviewed eScholarship.org Powered by the California Digital Library University of California Research JAMA | Original Investigation Association of Genetic Variants With Primary Open-Angle Glaucoma Among Individuals With African Ancestry The Genetics of Glaucoma in People of African Descent (GGLAD) Consortium Related article page 1705 IMPORTANCE Primary open-angle glaucoma presents with increased prevalence and a higher Supplemental content degree of clinical severity in populations of African ancestry compared with European or Asian ancestry. Despite this, individuals of African ancestry remain understudied in genomic research for blinding disorders. OBJECTIVES To perform a genome-wide association study (GWAS) of African ancestry populations and evaluate potential mechanisms of pathogenesis for loci associated with primary open-angle glaucoma. DESIGN, SETTINGS, AND PARTICIPANTS A 2-stage GWAS with a discovery data set of 2320 individuals with primary open-angle glaucoma and 2121 control individuals without primary open-angle glaucoma. The validation stage included an additional 6937 affected individuals and 14 917 unaffected individuals using multicenter clinic- and population-based participant recruitment approaches. Study participants were recruited from Ghana, Nigeria, South Africa, the United States, Tanzania, Britain, Cameroon, Saudi Arabia, Brazil, the Democratic Republic of the Congo, Morocco, Peru, and Mali from 2003 to 2018. -

APBB2 Antibody Cat
APBB2 Antibody Cat. No.: 27-061 APBB2 Antibody Specifications HOST SPECIES: Rabbit SPECIES REACTIVITY: Human, Mouse, Rat Antibody produced in rabbits immunized with a synthetic peptide corresponding a region IMMUNOGEN: of human APBB2. TESTED APPLICATIONS: ELISA, WB APBB2 antibody can be used for detection of APBB2 by ELISA at 1:1562500. APBB2 APPLICATIONS: antibody can be used for detection of APBB2 by western blot at 1 μg/mL, and HRP conjugated secondary antibody should be diluted 1:50,000 - 100,000. POSITIVE CONTROL: 1) Cat. No. XBL-10410 - Fetal Lung Tissue Lysate PREDICTED MOLECULAR 81 kDa WEIGHT: Properties PURIFICATION: Antibody is purified by peptide affinity chromatography method. CLONALITY: Polyclonal CONJUGATE: Unconjugated PHYSICAL STATE: Liquid September 25, 2021 1 https://www.prosci-inc.com/apbb2-antibody-27-061.html Purified antibody supplied in 1x PBS buffer with 0.09% (w/v) sodium azide and 2% BUFFER: sucrose. CONCENTRATION: batch dependent For short periods of storage (days) store at 4˚C. For longer periods of storage, store STORAGE CONDITIONS: APBB2 antibody at -20˚C. As with any antibody avoid repeat freeze-thaw cycles. Additional Info OFFICIAL SYMBOL: APBB2 ALTERNATE NAMES: APBB2, FE65L, FE65L1, MGC35575 ACCESSION NO.: NP_775098 PROTEIN GI NO.: 50083291 GENE ID: 323 USER NOTE: Optimal dilutions for each application to be determined by the researcher. Background and References APBB2 may modulate the internalization of beta-amyloid precursor protein.The protein encoded by this gene interacts with the cytoplasmic domains of amyloid beta (A4) BACKGROUND: precursor protein and amyloid beta (A4) precursor-like protein 2. This protein contains two phosphotyrosine binding (PTB) domains, which are thought to function in signal transduction. -

APBB2 (NM 001166051) Human Tagged ORF Clone Product Data
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for RC228759 APBB2 (NM_001166051) Human Tagged ORF Clone Product data: Product Type: Expression Plasmids Product Name: APBB2 (NM_001166051) Human Tagged ORF Clone Tag: Myc-DDK Symbol: APBB2 Synonyms: FE65L; FE65L1 Vector: pCMV6-Entry (PS100001) E. coli Selection: Kanamycin (25 ug/mL) Cell Selection: Neomycin ORF Nucleotide >RC228759 representing NM_001166051 Sequence: Red=Cloning site Blue=ORF Green=Tags(s) TTTTGTAATACGACTCACTATAGGGCGGCCGGGAATTCGTCGACTGGATCCGGTACCGAGGAGATCTGCC GCCGCGATCGCC ATGGCTGAACGGAAGAATGCCAAAGCGCTGGCCTGCAGCTCCTTACAGGAAAGGGCCAATGTGAACCTCG ATGTCCCTTTGCAAGTAGATTTTCCAACACCAAAGACTGAGCTGGTCCAGAAGTTCCACGTGCAGTACTT GGGCATGTTACCTGTAGACAAACCAGTCGGAATGGATATTTTGAACAGTGCCATAGAAAATCTTATGACC TCATCCAACAAGGAGGACTGGCTGTCAGTGAACATGAACGTGGCTGATGCCACTGTGACTGTCATCAGTG AAAAGAATGAAGAGGAAGTCTTAGTGGAATGTCGTGTGCGATTCCTGTCCTTCATGGGTGTTGGGAAGGA CGTCCACACATTTGCCTTCATCATGGACACGGGGAACCAGCGCTTTGAGTGCCACGTTTTCTGGTGCGAG CCTAATGCTGGTAACGTGTCTGAGGCGGTGCAGGCCGCCTGCATGTTACGATATCAGAAGTGCTTGGTAG CCAGGCCGCCTTCTCAGAAAGTTCGACCACCTCCACCGCCAGCAGATTCAGTAACCAGAAGAGTCACAAC CAATGTAAAACGAGGGGTCTTATCCCTCATTGACACTTTGAAACAGAAACGCCCTGTCACCGAAATGCCA ACGCGTACGCGGCCGCTCGAGCAGAAACTCATCTCAGAAGAGGATCTGGCAGCAAATGATATCCTGGATT ACAAGGATGACGACGATAAGGTTTAA Protein Sequence: >RC228759 representing NM_001166051 Red=Cloning site Green=Tags(s) MAERKNAKALACSSLQERANVNLDVPLQVDFPTPKTELVQKFHVQYLGMLPVDKPVGMDILNSAIENLMT