Evidence for an Early Innate Immune Response in the Motor Cortex of ALS Javier H
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Medical Study Center
https://www.facebook.com/Medicalstudycenter2012 https://www.facebook.com/Medicalstudycenter2012 Notes On CNS Physiology Gray Matter of Spinal Cord: in the spinal cord gray matter is in the form of H shaped pillars which can be divided into three types of columns i.e. anterior horn or ventral horn, posterior horn or dorsal horn and in segments from T1 to L2 there is lateral horn. Neurons in these horns are Ventral Horn: Two groups of neurons Alpha motor neurons which are large multi polar neurons and their nerve fibers are alpha efferents which innervate skeletal muscle. Gamma neurons present in ventral horn are small and multi polar neurons and there nerve fibers are gamma efferents which innervate the intrafusal fibers of the muscle spindles. Both these alpha and gamma efferents come out of the spinal cord through ventral root of spinal nerves. Dorsal Horn: there are 4 groups of neurons 1. Substantia gelatinosa: this group of neurons is present at the apex of the posterior gray column. These neurons receive afferent nerve fibers carrying impulses of pain, temperature and crude touch. 2. Nucleus Proprius: this group is located anterior to the first group. These neurons receive afferent nerve fibers carrying impulses of proprioception and two point tactile discrimination. 3. Clarke’s column or nucleus dorsalis: this group is present at the base of the posterior gray column. These neurons are present in segments from T1 to L3, 4. These neurons are part of spinocerebellar tract and these receive afferent nerve fibers from spinocerebellar tract. 4. Visceral Afferent Nucleus: Present at the base of the posterior horn lateral to the clarke’s column. -

Acupuncture and Pain Management
IN-DEPTH: INTEGRATIVE MEDICINE (COMPLEMENTARY & ALTERNATIVE MEDICINE) Acupuncture and Pain Management James D. Kenney, DVM There is a large and expanding body of scientific evidence supporting the use of acupuncture in pain management. In the last decade, our understanding of how the brain processes acupuncture anal- gesia has undergone considerable development. Profound studies on neural mechanisms underlying acupuncture analgesia have evolved rapidly and predominately focus on cellular and molecular substrate and functional brain imaging. The currently understood mechanisms of acupuncture analgesia are complex and involve direct and indirect neurohumoral effects that block pain percep- tion, reduce the pain response, relieve muscle spasms, and reduce inflammation. The analgesic mechanisms of acupuncture involve the spinal cord grey matter, hypothalamic-pituitary axis, mid- brain periaqueductal grey matter, medulla oblongata, limbic system, cerebral cortex, and autonomic nervous system. Electroacupuncture (EA) stimulation of these sites results in activation of descend- ing pathways that inhibit pain through endogenous opioid, noradrenergic, and serotonergic sys- tems. There are growing numbers of human trials supporting the use of acupuncture as an evidence- based practice for pain management in human medicine. There are many studies that support the efficacy of acupuncture for low back pain, neck pain, chronic idiopathic and migraine headaches, knee pain, shoulder pain, fibromyalgia, temporomandibular joint pain, and postoperative pain. Although the number of well-designed, controlled clinical research studies in veterinary medicine is lagging behind the number of studies in human medicine, much of the basic science research has been done in animals with neurophysiology that is more similar to veterinary patients than humans. Although there is research to support EA as an evidence-based practice for the control of back pain in horses, additional studies are needed in other clinical situations in veterinary medicine where pain manage- ment is required. -
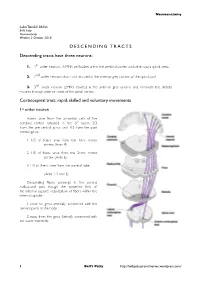
DESCENDING TRACTS Descending Tracts Have Three Neurons
Neuroanatomy Luka Tomšič Ahčin Bell’s Palsy Neuroanatomy Written: 2 October 2010 DESCENDING TRACTS Descending tracts have three neurons: 1. 1st order neurons (UMN): cell bodies are in the cerebral cortex and other supra spinal areas 2. 2nd order neurons: short and situated in the anterior grey column of the spinal cord 3. 3rd order neuron (LMN): situated in the anterior grey column and innervate the skeletal muscles through anterior roots of the spinal nerves Corticospinal tract: rapid, skilled and voluntary movements 1st order neuron Axons arise from the pyramidal cells of the cerebral cortex (situated in the 5th layer), 2/3 from the pre central gyrus and 1/3 from the post central gyrus: 1. 1/3 of fibers arise from the 1stry motor !!cortex (Area 4) 2. 1/3 of fibers arise from the 2ndry motor !!cortex (Area 6) 3. 1/3 of fibers arise from the parietal lobe ! (Area 1, 2 and 3). Descending fibers converge in the corona radiata and pass though the posterior limb of the internal capsule; organization of fibers within the internal capsule: 1. close to genu (medial): concerned with the cervical parts of the body 2. away from the genu (lateral): concerned with the lower extremity. 1! Bell’s Palsy http://bellspalsycranialnerves.wordpress.com/ Neuroanatomy The tract then passes through the middle 3/5 of the basis pedunculi of the midbrain; organization of fibers in the midbrain: 1. medially: cervical parts of the body 2. laterally: lower limbs. When the tract enters the pons, it's broken into many bundles by the transverse pontocerebellar fibers. -

Gen Anat-Nervous System
Nervous system • Controlling & Coordinating System Functions • Regulates all activity (Voluntary & Involuntary) • Adjust Acc. to changing external and internal environment Nervous System Subdivisions CNS (Central Nervous System) PNS( Peripheral Nervous System) ANS (Autonomic Nervous system CNS Brain ( Encephalon) Spinal Cord ( Sp. Medulla) Parts • Cerebrum • Cerebellum •Brain Stem -Mid Brain -Pons -Medulla PNS ( Peripheral Nervous system) Two Components 1. Somatic ( Cerebrospinal) ---12 Pair Cranial Nerves ----31 pair Spinal Nerves 2. Visceral ( Autonomic Nervous System – ANS) ----Visceral or Splanchnic nerves two – subdivisions i) Sympathetic ii) Parasympathetic Somatic Component • Deals with any change in external environment – Extroceptive or Proprioceptive General Sensations like • Pain , Touch , Temp. --- From Skin • Sensations from muscles , bones , joints, limbs Special Sensations like • Vision • Hearing • Balancing – Through vestibular receptors Cranial Nerves 1. OLFACTORY 7. FACIAL 2. OPTIC 8. VESTIBULO-COCLEAR 3. OCCULOMOTOR 9. GLOSSOPHARYNGEAL 4. TROCHLEAR 10. VAGUS 5. TRIGERMINAL 11. ACCESSORY 6. ABDUCENT 12. HYPOGLOSSAL 31 Pairs Spinal Nerves Includes Cervical -8 (C1 ----C8) Thoracic -12 (T1-T12) Lumbar – 5 (L1-L5) Sacral - 5 (S1– S5) Coccyx – 1 (Co -1) Spinal Nerve Joining of ant. and Post. nerve roots Spinal Segment Length of the spinal cord originating rootlets of one spinal nerve Spinal Nerve Dorsal Root & Ventral Root Join to form trunk of spinal nerve At intervertebral foramina divide into Dorsal and ventral ramus Dorsal ramus runs posteriorly and divide in Medial and Lateral Branches to supply muscles of back, and give cut. Branches Ventral ramus runs anteriorly and give lateral cutaneous branch which further subdivide into: Ant and post branches Rest continue as ant. cutaneous Br. Ventral Rami of Cervical, Lumbar. -

(Neuron) (Spinal Cord and Brain) Neuroglia (Support) Neuroglia
210 اوم څپرکې عصبي نسج (NERVOUS TISSUE) (impulse) (neuron) (spinal cord and brain) neuroglia (support) neuroglia ٧- ١ شکل دماغ 1012 211 اوم څپرکې عصبي نسج (axons) (NEURON STRUCTURE) cell body Cell body perikaryon Soma ٧-٢ شکل دنيوران جوړښت centrioles granules basal body nissl substance 212 اوم څپرکې عصبي نسج Endoplasmic Reticulum neurotransmitters neurofibrils microtubules microfilaments neurofibrils microtubules substantia (pigmented granules) nerumelanin nigra lipofuscin cell body neuritis Dendrites Axons cell body Dendrites nissle dendrites dendrites granules dendrites cell body 213 اوم څپرکې عصبي نسج axon nissle granuls (axon and dendrites) axon cell body dendrites cell body MAP-2 dendrites dendrites nissl granules axon cell body cell body initial segments axon hillock hillock Schwann cells (sheath) oligodendrocytes Schwann cells Schwann cell ٧- ٤ شکل Schwann cells mesaxon 214 اوم څپرکې عصبي نسج (mesaxon) mesaxon myelin sheath schwann cells myelin sheath neurolimmal sheath neurilemma Schwann cell sheath myelinated axons (myelin sheath) axons Schwann cells myelin sheath myelin 215 اوم څپرکې عصبي نسج node of Ranvier myelin sheath (nodes of Ranvier) (Internode) unmyelinated axons myelin sheath mesaxon Schwann cells Schwann cell body collaterals telodendria boutons terminal boutons of terminaux synapse ganglions synapse 216 اوم څپرکې عصبي نسج dendrites 6,8,3 (nerve fibers) cell body cell bodies cell bodies 120µm cell body 5µm ٧-٨ شکل داکسون او cell body نيوران ارتبات multipolar neurons (bipolar neuron) unipolar neuron bipolar neuron (pseudounipolar -

Upper Motor Neuron and Lower Motor Neuron Lesions
Lecture XXIV: Upper Motor Neuron and Lower Motor Neuron Lesions EDITING FILE GSLIDES IMPORTANT MALE SLIDES EXTRA FEMALE SLIDES LECTURER’S NOTES 1 UPPER MOTOR NEURON AND LOWER MOTOR NEURON LESIONS Lecture 24 OBJECTIVES - Describe the functional anatomy of upper and lower motor neurons. - Describe the functional anatomy of upper and lower motor neurons. - Explain features of Brown Sequard Syndrome. - Correlate the site of lesion with pattern of loss of sensations. - Describe facial, bulbar and pseudobulbar palsy. Facts about Spinal Cord ● Embryological development — growth of cord lags behind—mature spinal cords ends at L1. ● It has 31 segments. Upper & Lower motor neuron UMNs control LMNs through two different pathways: ● Pyramidal system. ● Extrapyramidal system. Figure 24-1 Figure 24-2 Pure corticospinal tract lesion cause hypotonia instead of spasticity. The reason is that pure pyramidal tract lesion is very very rare, and spasticity is due to loss of inhibitory control of extrapyramidal tract. Figure 24-3 Figure 24-4 Pure corticospinal tract lesion cause hypotonia instead of spasticity. The reason is that pure pyramidal tract lesion is very very rare, and spasticity is due to loss of inhibitory control of extrapyramidal tract. 2 UPPER MOTOR NEURON AND LOWER MOTOR NEURON LESIONS Lecture 24 Internal Capsule Lenticulostriate arteries from the - Reciprocal connections between thalamus middle cerebral artery and cortex are found in four limbs of the internal capsule: 1. Anterior limb with frontal lobe. 2. Posterior limb with parietal lobe. 3. Retrolenticular limb with occipital lobe. 4. Sublenticular limb with temporal lobe. - The genu contains the corticobulbar Axons. - Corticospinal axons are in the posterior limb. -

Histological Changes in the Central Nervous System of Swine 1 Inoculated with Some Enteroviruses )
Acta vet. scand. 1963, 4, 395-403. From the Departments of Medicine and Anatomy and Histology, Royal Veterinary College, Stockholm. HISTOLOGICAL CHANGES IN THE CENTRAL NERVOUS SYSTEM OF SWINE 1 INOCULATED WITH SOME ENTEROVIRUSES ) By M. Sibalin and G.Winqvist The Teschen virus strains, which belong to the enteroviruses, are known since long to cause poIioencephalomyelitis in pigs. All Teschen strains comprise one serotype, which according to Mayr (8) can be divided into two sub-types. The causative agent of the Talfan disease in England (5) and possibly also the benign enzootic paresis in Denmark (14) are Teschen viruses (2). The histological changes in the CNS at Teschen disease have been described in detail (4, 7, 14). Investigations on swine enteroviruses, which do not cross neutralize with the Teschen strains but show similar clinical and morphological pictures in experimental pigs, have been re ported by Betts & Jennings (1), Sibalin & Lannek (10), and Izawa et al. (6) . This report deals with histological changes observed in the CNS of colostrum-deprived piglets used in pathogenicity tests of enteroviruses isolated in Sweden (10, 12, 13). MATERIAL AND METHODS 1. Virus strains. The viruses used in the experimental work have been described and characterized as enteroviruses (10, 11, 12, 13). All strains, except S180/4 and U6, are shown in Table 1. 1) Aided by grants from Fonden for framjande av forsknings- och forsoksverksarnheten p:\ jordbrukets omrade. Table 1. Degree and localization of the histologic changes in the eNS. 1:1 Ino culum 0 :l c " 0 :; ;: etJ";' .. 0 til c; ..s e;... bIJ 1:1 a:> . -

Serotonergic Neurons of the Nucleus Raphe Magnus in the Control Of
Serotonergic neurons of the nucleus raphe Magnus in the control of nociceptive transmission in the dorsal horn of the spinal cord : an optogenetic study in different pathophysiological contexts Franck Aby To cite this version: Franck Aby. Serotonergic neurons of the nucleus raphe Magnus in the control of nociceptive transmis- sion in the dorsal horn of the spinal cord : an optogenetic study in different pathophysiological contexts. Neurons and Cognition [q-bio.NC]. Université de Bordeaux, 2019. English. NNT : 2019BORD0354. tel-02491370 HAL Id: tel-02491370 https://tel.archives-ouvertes.fr/tel-02491370 Submitted on 26 Feb 2020 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. THÈSE PRÉSENTÉE POUR OBTENIR LE GRADE DE DOCTEUR DE L’UNIVERSITÉ DE BORDEAUX Ecole Doctorale des sciences de la vie et de la santé Spécialité Neurosciences Par Franck Aby Les neurones sérotoninergiques du noyau raphé Magnus dans le contrôle de la transmission nociceptive dans la corne dorsale de la moelle épinière : une étude optogénétique dans différents contextes pathophysiologiques Sous la direction de : Pr Pascal Fossat Soutenue le 13 Décembre 2019 Membres du Jury Mme. Nadjar, Agnès Professeur NutriNeuro Bordeaux Présidente Mme. -

CNS)- the Brain and Spinal Cord
Introduction Remember studying Nervous Tissue as a Basic Tissue? 1. Epithelial tissue 2. Connective tissues 3. Muscle tissue 4. Nervous tissue Well, during this lab you will review these tissues as observed in both the: i. Central Nervous System (CNS)- the brain and spinal cord. ii. Peripheral Nervous System (PNS) - peripheral nerves/ganglia outside the CNS. Learning objectives and activities Using the Virtual Slidebox: A Recognize nervous tissue in histological sections and discuss the organization/function of the cells present. B Examine the features of peripheral nerves in both longitudinal and cross section that allow you to identify them in tissue sections. C Differentiate between sensory and autonomic (sympathetic/parasympathetic) ganglia. D Identify the different functional regions and cells of the spinal cord. E Identify the major layers and cells of the cerebellum ad the cerebral cortex. F Complete the self-quiz to test your understanding and master your learning. Despite its complexities, the nervous system is composed entirely of two cell types: a network of nerve cells (neurons) that transmit signals throughout the nervous system and the glial cells that support them. Neurons Examine Slide 1 (16) to investigate the basic structure of a neuron ii i. Cell body (perikaryon). Most cell bodies are located in the central nervous system (brain and spinal cord) or in ganglia of the peripheral nervous i system. They contain darkly stained granules called Nissl bodies which are the ribosomes used to manufacture iii neurotransmitter in the cell. iv ii. Dendrites Dendrites extend from the cell body. These are short, highly branched structures that receive nerve impulses from other neurons. -

Neurophysiology
Neurophysiology Second edition Copyright StudyAid 2018 Authors Kamil Winnicki Amalie Misund Illustrations Amalie Misund Nora Sønstebø Editor Amalie Misund Booklet Disclaimer All rights reserved. No part of this book may be reproduced in any form on by an electronic or mechanical means, without permission from StudyAid. Although the authors have made every effort to ensure the information in the booklet was correct at date of publishing, the authors do not assume and hereby disclaim any liability to any part for any information that is omitted or possible errors. The material is taken from a variety of academic sources as well as physiology lecturers, but are further incorporated and summarized in an original manner. It is important to note, the material has not been approved by professors of physiology. All illustrations in the booklet are original. This booklet is made especially for students at the Jagiellonian University in Krakow by tutors in the StudyAid group (students at JU). It is available as a PDF and is available for printing. If you have any questions concerning copyrights of the booklet please contact [email protected]. About StudyAid StudyAid is a student organization at the Jagiellonian University in Krakow. Throughout the academic year we host seminars in the major theoretical subjects: anatomy, physiology, biochemistry, immunology, pathophysiology, supplementing the lectures provided by the university. We are a group of 25 tutors, who are students at JU, each with their own field of specialty. To make our seminars as useful and relevant as possible, we teach in an interactive manner often using drawings and diagrams to help students remember the concepts. -

From the Poliomyelitis Research Center, Department of Epidemiology, Johns Hopkins University, Baltimore)
CYTOCHROME OXIDASE IN NORMAL AND REGENERATING NEURONS* BY HOWARD A. HOWE, M.D., Am) ROBERT C. MELLORS, M. D. (From the Poliomyelitis Research Center, Department of Epidemiology, Johns Hopkins University, Baltimore) (Received for publication, January 30, 1945) The observation has been made that the motor cells of the monkey's spinal cord become resistant to destruction by poliomyelitis virus during most of the period of regeneration which follows the severance of a peripheral nerve (1, 2). This phenomenon, which can be easily induced experimentally, appears during the 3rd week of nerve regeneration and persists until functional recovery has been effected. The motor neuron, therefore, offers a situation which is unique in providing an opportunity for the study of some of the factors which make for cellular resistance, and it should be possible by chemical means, as well as by ordinary histological techniques, t3 detect some differences, either qualitative or quantitative, between normally susceptible tissue and that which has been rendered resistant. Chemical studies of nervous tissue have dealt largely with the processes of aerobic and anaerobic respiration. They have been limited in scope by a number of factors, for example, the fragility of the tissue, the admixture of grey and white matter which have divergent metabolic rates, the difficulty of measuring the activities of the individ- ual groups of cells and of correlating them with different functional types or states. Efforts have been made by a few investigators to study different functional conditions in complex mixtures of central nervous tissue. One of the most successful of these is that of F]exner, Flexner, and Straus (3) who have been able to make a correlation between cytochrome-cytochrome oxidase activity and morphological differentiation in the embryonic nervous system. -

Dissociate” Responses
Introduction to Neural Transmission and the Neural Mechanisms of Acupuncture Analgesia Taylor C. Thorne University of North Carolina Wilmington 2016 People Try Acupuncture Therapy The Neuron and Transmission of Information Outline of Neural Transmission 1. Introduction of the Neuron 2. Anatomy of the Neuron 3. Classification of Neurons - structural - functional 4. The Electrical/Chemical Nature of the Neuron 5. Putative Neurotransmitters 6. Classical Neurotransmitters - effects of neurotransmitters 7. Postsynaptic Receptor Types 8. Activity at the Synapse 9. Antagonistic vs. Agonistic 10.Overview: Components/Functions of the Nervous System 11.The Peripheral Nervous System - somatic nervous system To better understand the process in which acupuncture - autonomic nervous system functions it is important to first grasp the understanding - sympathetic nervous system of the nature of neural transmission (aka communication - parasympathetic nervous system between neurons) within/between the central nervous - autonomic nervous supply to organs system (CNS) and peripheral nervous system (PNS) - nerves and plexuses 12.The Central Nervous System 13.Neurotransmitter Systems In the Brain - norepinephrine - dopamine - serotonin - acetylcholine 14.Brain Pain Pathways Introduction: The Neuron Facts Function • size/length • processes/transmits information via electrical- • sensory neurons can have axons that extend from chemical signals toes to the posterior column of the spinal cord • communication between neurons occurs via • over 1.5 meters in length