Ucd Chem 124L: Lab Manual
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Laboratory Glassware N Edition No
Laboratory Glassware n Edition No. 2 n Index Introduction 3 Ground joint glassware 13 Volumetric glassware 53 General laboratory glassware 65 Alphabetical index 76 Índice alfabético 77 Index Reference index 78 [email protected] Scharlau has been in the scientific glassware business for over 15 years Until now Scharlab S.L. had limited its sales to the Spanish market. However, now, coinciding with the inauguration of the new workshop next to our warehouse in Sentmenat, we are ready to export our scientific glassware to other countries. Standard and made to order Products for which there is regular demand are produced in larger Scharlau glassware quantities and then stocked for almost immediate supply. Other products are either manufactured directly from glass tubing or are constructed from a number of semi-finished products. Quality Even today, scientific glassblowing remains a highly skilled hand craft and the quality of glassware depends on the skill of each blower. Careful selection of the raw glass ensures that our final products are free from imperfections such as air lines, scratches and stones. You will be able to judge for yourself the workmanship of our glassware products. Safety All our glassware is annealed and made stress free to avoid breakage. Fax: +34 93 715 67 25 Scharlab The Lab Sourcing Group 3 www.scharlab.com Glassware Scharlau glassware is made from borosilicate glass that meets the specifications of the following standards: BS ISO 3585, DIN 12217 Type 3.3 Borosilicate glass ASTM E-438 Type 1 Class A Borosilicate glass US Pharmacopoeia Type 1 Borosilicate glass European Pharmacopoeia Type 1 Glass The typical chemical composition of our borosilicate glass is as follows: O Si 2 81% B2O3 13% Na2O 4% Al2O3 2% Glass is an inorganic substance that on cooling becomes rigid without crystallising and therefore it has no melting point as such. -

Site-Directed Spin Labeling of Large Riboswitches Using Click Chemistry
Site-Directed Spin Labeling of Large Riboswitches Using Click Chemistry Dissertation zur Erlangung des Doktorgrades (Dr. rer. nat.) der Mathematisch-Naturwissenschaftlichen Fakultät der Rheinischen Friedrich-Wilhelms-Universität Bonn vorgelegt von Mark Kerzhner aus Winnyzja, Ukraine Bonn 2018 Angefertigt mit Genehmigung der Mathematisch-Naturwissenschaftlichen Fakultät der Rheinischen Friedrich-Wilhelms-Universität Bonn 1. Gutachter: Prof. Dr. Michael Famulok 2. Gutachter: Prof. Dr. Olav Schiemann Tag der Promotion: 25.10.2018 Erscheinungsjahr: 2019 Parts of this thesis have been published in: Kerzhner, M.; Abdullin, D.; Więcek, J.; Matsuoka, H.; Hagelueken, G.; Famulok, M.; Schiemann, O., Post-synthetic Spin-Labeling of RNA through Click Chemistry for PELDOR Measurments, Chem. Eur.J. 2016, 22, 12113 –12121. Kerzhner, M.; Matsuoka, H.; Wuebben, C.; Famulok, M.; Schiemann, O., High- Yield Spin Labeling of Long RNAs for Electron Paramagnetic Resonance Spectroscopy, Biochemistry, 2018, 57, 2923–2931. Danksagung An erster Stelle möchte ich Herrn Prof. Dr. Michael Famulok danken, dass er mir die Chance ermöglicht hat, an diesem anspruchsvollen aber hochinteressanten Projekt zu arbeiten. Außerdem bin ich ihm sehr dankbar, dass er mir über die Jahre sein Vertrauen geschenkt hat. Großer Dank gilt außerdem dem Kooperationspartner Herrn Prof. Dr. Olav Schiemann, ohne dessen Unterstützung die Umsetzung dieses Projektes nicht möglich gewesen wäre. Er und seine Arbeitsgruppe haben entscheidende Beiträge zu den erzielten Ergebnissen dieser Arbeit geleistet. Allen Mitgliedern meiner Prüfungskommission danke ich für die Begutachtung dieser Dissertation. Ich danke meinen Kooperationspartnern aus der Arbeitsgruppe von Herrn Prof. Dr. Olav Schiemann. Bei Dr. Dinar Abdullin, Dr. Hideto Matsuoka und Dr. Andreas Meyer bedanke ich mich herzlich für die zahlreichen PELDOR-Messungen sowie Hilfe bei verschiedenen Fragestellungen. -

Viruses at Glycocalyx Barriers SI Bioarxiv
Glycocalyx crowding with synthetic mucin mimetics strengthens interactions between soluble and virus-associated lectins and cell surface glycan receptors Authors: aDaniel J. Honigfort, b, † Meghan O. Altman, bPascal Gagneux, and Kamil Godula*,a Author Affiliation: aDepartment of Chemistry and Biochemistry, University of California San Diego, 9500 Gilman Drive, La Jolla, CA 92093-0358, USA bDepartment of Pathology, University of California San Diego, 9500 Gilman Drive, La Jolla, CA 92093-0687, USA Supporting Information 1 Table of contents: Instrumentation and reagents. ............................................................................................................ 4 Synthesis of propargyl glycosides. ........................................................... Error! Bookmark not defined. Scheme S1. Preparation of β-propargyl glycosides via the Schmidt glycosylation.Error! Bookmark not defined. Synthesis of glycopolymers GP-S/M/L ............................................................................................... 5 Scheme S2. Synthesis of Glycopolymers through iterative CuAAc click strategy. ................................ 5 General Procedure for the preparation of poly(epichlorohydrin) (P1). ............................................ 5 General Procedure for the end-functionalization of poly(epichlorohydrin) (P2). ............................ 6 General Procedure for the preparation of poly(Glycidyl Azide) (P3). .............................................. 6 General procedure for the preparation of Glycopolymers -

Chem 1140; Techniques for Handling Air-Sensitive Compounds
P. Wipf 1 Chem 1140 Chem 1140; Techniques for Handling Air-Sensitive Compounds • Introduction • The Glove Box • Schlenk Techniques • Drying and Degassing Solvents What are Air-Sensitive Compounds? Materials which oxidize, decompose or even explode under the influence of oxygen or moisture. • Pyrophoric Compounds Metal alkyls and aryls e.g. RMgX, RLi, RNa,R3Al, R2Zn Metal carbonyls e.g. Ni(CO)4, Fe(CO)5, Co2(CO)8 Alkali metals e.g. Na, K, Cs Metal powders e.g. Al, Co, Fe, Mg, Pd, Pt, Zn Metal hydrides e.g. NaH, KH, LiAlH4 Hydrides e.g. B2H6, PH3, AsH3 Boranes, phosphines, arsenes, etc. e.g. Et3B, R3P, R3As • Chemicals which react violently with water Metal hydrides, metal amides (NaNH2), metal alkyls and aryls, metals, metal powders, hydrides, many main group halides (BCl3, BF3, AlCl3, PCl3, SiCl4), inorganic acid halides (POCl3, SOCl2), low molecular weight organic acid halides and anhydrides. Glove boxes and Schlenk techniques do NOT protect from explosive or shock sensitive materials or mixtures!!! Also, they only provide limited protection from toxic compounds. P. Wipf 2 Chem 1140 The Glove Box • The best way to keep things away from atmospheric oxygen and water is to work in a fully enclosed “bench top,” containing an “inert atmosphere,” which one could reach into with gloves. Such a device is called a “glove box” or a “dry box”. There are also cheap “glove bags”, bags you can fill with inert gas and reach into with attached gloves. The Glove Box A glove box has four important components: 1. The actual “box” is a large aluminum chamber with a plastic front window and two impressive looking gloves. -

A Smartphone Biosensor Based on Analysing Structural Colour of Porous Silicon† Cite This: Analyst, 2019, 144, 3942 Tengfei Cao, a Yiliang Zhao, a Crystal A
Analyst View Article Online PAPER View Journal | View Issue A smartphone biosensor based on analysing structural colour of porous silicon† Cite this: Analyst, 2019, 144, 3942 Tengfei Cao, a Yiliang Zhao, a Crystal A. Nattoo, b,c Rabeb Layouni d and Sharon M. Weiss *a,c We report a smartphone compatible, low-cost porous silicon biosensor, which correlates the structural colour of a porous silicon microcavity (PSiM) to spectral peak position. Molecules captured in the PSiM cause a colour change that can be quantified through image analysis. Minimal external accessories are employed. Spectrometer measurements of the PSiM reflectance spectrum shifts are carried out concur- rently with the smartphone measurements to benchmark the accuracy of the smartphone biosensor. We Received 4th January 2019, estimate that the smartphone biosensor supports an equivalent accuracy of 0.33 nm for the detection of Accepted 5th May 2019 colour changes corresponding to spectral shifts of the PSiM. Biosensing functionality is demonstrated DOI: 10.1039/c9an00022d using a biotin–streptavidin assay with an estimated detection limit of 500 nM. The PSiM-smartphone bio- rsc.li/analyst sensor is a promising platform for label-free point of care diagnostics. 1. Introduction designed for a single purpose instead of multi-parameter testing. Moreover, the data processing capabilities of these Point-of-care (POC) testing is a fast-growing sector in the sensors are usually basic and not intended for more compli- diagnostics market due to the convenience and low cost of cated detection and analysis tasks. Over the past few years, POC measurements.1 Biosensors are the crucial elements of smartphones have emerged as a promising host platform for POC diagnostics, which transform biomolecular or medical POC diagnostics systems due to their ubiquitous availability information into easily understandable readouts. -

Standard Operating Procedures Standard Operating Procedures Tolman Laboratory
Tolman Group Standard Operating Procedures Standard Operating Procedures Tolman Laboratory v.7 updated May 2016 The intent of this manual is to inform you, the chemist, of the potential hazards that exist in the laboratory. This is by no means a manual to memorize or a manual of instruction. These procedures are not intended to be exhaustive or to serve as a single method of training. The procedures outlined here are meant as a refresher for those who have already received training by senior lab members. If in doubt of any procedure, please ask someone before carrying out said procedure. Asking for help when in doubt is the responsibility of every lab member. The first time you use all equipment and perform new experimental procedures, you should have someone work with you to teach you and to inform you of potential hazards, and to help prevent accidents. The purpose of this manual is to supplement personal one-on-one training with safety information that will protect you personally, as well as other lab members. It is important to make sure that you minimize your exposure to chemicals and decrease the chances of any accidents. By reviewing (and later referring to) this manual, you can be aware of the potential hazards in the lab and learn careful techniques. It is your responsibility to learn about safety and proper handling of chemicals from the resources available, including your laboratory members, other persons in the department, and the Chemical Hygiene Plan, which can be accessed via the web, from the Safety link on the Chemistry Webpage www.chem.umn.edu, or http://www.chem.umn.edu/services/safety/. -

Li Deposited on Licl: an Efficient Reducing Agent
Electronic Supplementary Material (ESI) for New Journal of Chemistry. This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2020 Li deposited on LiCl: An Efficient Reducing Agent Xiaojun Su, Leif E. Laperriere and Caleb D. Martin* Baylor University, Department of Chemistry and Biochemistry, One Bear Place #97348, Waco, TX 76798 E-mail: [email protected] -Supporting Information- 1 Table of Contents General Details p 3 Normality test of lithium granule weight p 4 Experimental Procedures p 5 Reaction Yields with Different Li Sources p 6 SEM of LiCl before grind p 7 SEM of LiCl after grind p 8 SEM of ca. 5% w/w Li/LiCl powders p 10 2 General Details. All manipulations were performed under an inert argon atmosphere using standard Schlenk techniques or in a MBraun Unilab glovebox. Solvents were purchased from commercial sources as anhydrous grade, dried further using a JC Meyer Solvent System with dual columns packed with solvent-appropriate drying agents. LiCl (99.5+%) was purchased from Fisher Chemical, Lithium granules (99.0+%) were purchased from Acros Organics, Diphenylacetylene (99.0%) was purchased from Alfa Aesar, Dimethyltin dichloride (99.0+%) was purchased from TCI America, Tetramethylsilane (TMS, 99.9+%) was purchased from Sigma-Aldrich, CDCl3 for NMR spectroscopy was purchased from Cambridge Isotope Laboratories and used as received. Multinuclear NMR spectra were recorded on a Bruker 400 MHz spectrometer. Origin Pro 8 was used for the data processing. Scanning electron microscopy (SEM) was recorded on a Tabletop Scanning Electron Microscope TM3030 Plus. An AICOOK blender (Model: NY-8608MA) was used to ground LiCl samples. -

Organic Chemistry Laboratory Methods
Organic chemistry – laboratory methods Organic chemistry Laboratory methods Bartosz Szyszko 1 Organic chemistry – laboratory methods Organic chemistry – laboratory methods Description This course deals with laboratory techniques in organic chemistry laboratory. Upon successful completion of this course students will posess practical skills required for work in modern chemical laboratory. Website http://do.chem.uni.wroc.pl/przedmiot/Metody-Laboratoryjne-laboratorium-ChOrg2 Language English Prerequisites basic organic chemistry Time commitment 90 hours; 5 preparatory sessions (30 hours) and 7 regular sessions (60 hours) Learning objectives 1. Purification and drying organic solvents and reagents by distillation in an inert and moisture-free atmosphere. 2. Microscale synthesis. 3. Running a reaction for a long time (also under reflux). 4. Synthesis under moisture- and air-free condtions. 5. Running a reaction at low temperature. 6. Performing multi-step synthesis without isolation of the intemediates. 7. Isolation of the product from a mixture containing very reactive reagents. 8. Isolation of the product by distillation, crystallization and chromatography. 9. Using Schlenk line. Removing solvents with high vacuum. Performing a vacuum distillation (also fractional) and bulb-to-bulb distillation. Calendar and class meetings Before start of the course all the students will participate in the fire-fighting training provided by specialized Fire-Fighters. Date of the training will be provided in near future. wednesday, 1100-1700 class date class date 1 2 XI 7 14 XII 2016 2 9 XI 8 4 I 2017 3 16 XI 9 11 I 2017 4 23 XI 10 18 I 2017 5 30 XI 11 25 I 2017 6 7 XII 12 1 II 2017 Instructors Dr. -

Supporting Information � Wiley-VCH 2014
Supporting Information Wiley-VCH 2014 69451 Weinheim, Germany Chemically Orthogonal Three-Patch Microparticles** Sahar Rahmani, Sampa Saha, Hakan Durmaz, Alessandro Donini, Asish C Misra, Jaewon Yoon, and Joerg Lahann* anie_201310727_sm_miscellaneous_information.pdf Table of Contents: Supplemental Section 1: Materials………………………………………...…………………………....................................………………..11 Supplemental Section 2: Experimental Section • 2.1: Polymerization, deprotection, and characterization of polymer 2……………...……..…………….……………………….…..12 • 2.2: Functionalization and characterization of polymers 3, 4, and 5………………………………………….……………..….…....13 • 2.3: Fabrication of multicompartmental microparticles, their surface modification & characterization………….…………………15 • 2.4: Selective surface functionalization of microparticles containing either polymer 1 or 2………………………………………...17 • 2.5: Full Raman spectra of the polymer 4 containing compartment……………..…………………………………..………….……18 • 2.6: Selective surface modification of microparticles containing polymers 2 and 5….....……………………………..…………… 19 • 2.7: Full Raman spectra of polymer 4 and 5 containing compartments……………………………………...………………………20 References: ………………………………………………………………………………..................................……………………………...21 Supplemental Section 1: Materials O-benzyl-L-serine was purchased from Alfa Aesar and diethyl ether was purchased from Acros Organics. Triethyl amine, bromoacetyl bromide, dimethyl amino pyridine, benzyl alcohol, l-lactide, tin (II) ethyl hexanoate, anhydrous toluene, palladium 10% wt. -

Schlenk Line Design and Safety by Patrick Frank Stanford Synchrotron Radiation Lightsource, SLAC, Stanford University 04 April 2011
Schlenk Line Design and Safety By Patrick Frank Stanford Synchrotron Radiation Lightsource, SLAC, Stanford University 04 April 2011 This report will begin with a description of a functioning Schlenk line, describing the separate parts and how they function together. Following after is a discussion of the various operational and physical (not chemical) elements of safety when using the Schlenk line. General PPE recommendations: • Safety glasses or goggles are required all the while a Schlenk line is in use. Face shields are optional. • Safety glasses are required when obtaining or using liquid nitrogen. • Thermal gloves are required as needed when dispensing or using liquid nitrogen or cold Dewars. • Lab coat and face shield are required if using especially reactive chemicals (pyrophorics or explosive mixtures). • Plastic lab coats are to be avoided unless they are explicitly rated as flame-proof. • The hood sash must be pulled down when the Schlenk line is unattended. • An explosion shield required when working with especially reactive chemicals (pyrophorics or explosive mixtures). Explosion hazard is evaluated on an experimental case-by-case basis. • An explosion shield is optional during vacuum distillation. I. Schlenk Line Description I.1 Introduction. Schlenk line is a tubular glass apparatus used to perform air-free bench- top chemical manipulations. Air-free work is required when reagents or products are sensitive to oxidation. A Schlenk line centrally consists of a duplex of glass tubes connected side-by-side, which together is called a “manifold.” The manifold is connected to a vacuum pump and a source of inert gas (N2 or Ar). A Schlenk line is most safely operated within a working hood. -
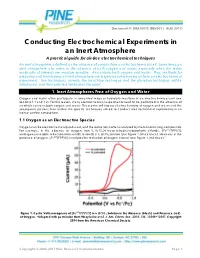
Conducting Electrochemical Experiments in an Inert Atmosphere
Document #: DRA10072 (REV001 | AUG 2017) Conducting Electrochemical Experiments in an Inert Atmosphere A practical guide for air-free electrochemical techniques An inert atmosphere is defined as the absence of oxygen from an electrochemical cell. Sometimes, an inert atmosphere also refers to the absence of both oxygen and water, especially when the redox molecules of interests are moisture sensitive. Air contains both oxygen and water. Thus, methods for preparing and maintaining an inert atmosphere are largely about removing air from an electrochemical experiment. Two techniques, namely, the benchtop technique and the glovebox technique, will be introduced, and their practical limitations discussed. 1. Inert Atmospheres Free of Oxygen and Water Oxygen and water often participate in unwanted redox or hydrolysis reactions in an electrochemical cell (see Sections 1.1 and 1.2). For this reason, many electrochemical experiments need to be performed in the absence of air which contains both oxygen and water. This section will discuss electrochemistry of oxygen and water and the subsequent sections then outline the specific techniques utilized to conduct electrochemical experiments in an inert or air-free atmosphere. 1.1 Oxygen as an Electroactive Species Oxygen can be electrochemically reduced, and the reduction is often catalyzed by metal-containing compounds. For example, in the absence of oxygen, iron 5, 10, 15, 20-meso-tetraphenylporphyrin chloride, [ ( ) ], undergoes reversible reduction from iron(III) to iron(II) in a solution (see Figure 1, black trace). However, in the presence of oxygen, [ ( ) ] catalyzes the reduction of oxygen instead (see Figure 1, red trace).1 4 Figure 1. Cyclic Voltammograms of [ ( ) ] in a Solution in the Absence (black) and Presence (red) of Oxygen. -

Hydrogen Cyanide Chemical Specific Standard Operating Procedure
Hydrogen Cyanide Chemical Specific Standard Operating Procedure Hydrogen Cyanide (HCN) H224 H300 H310 H330 H370 This SOP is not a substitute for hands-on training. Print a copy and insert into your laboratory SOP binder. Department: Chemistry Date SOP was written: Wednesday April 25, 2016 Date SOP was approved by PI/lab supervisor: Name: R. Sarpong Principal Investigator: Signature: ______________________________ Name: Melissa Hardy/Justin Jurczyk Internal Lab Safety Coordinator or Lab Manager: Lab Phone: 406-696-1225/412-728-1952 Office Phone: 510-642-6312 Name: Melissa Hardy/Justin Jurczyk Emergency Contact: Lab Phone: 406-696-1225/412-728-1952 Latimer Hall Location(s) covered by this SOP: 831,832,834,836,837,838,839,842,844,847,849 1. Purpose This SOP covers the precautions and safe handling procedures for the generation, use, and storage of Hydrogen Cyanide (HCN). If you have questions concerning the applicability of any recommendation or requirement listed in this procedure, contact the Principal Investigator/Laboratory Supervisor or the campus Chemical Hygiene Officer at [email protected]. 2. HCN Information Before working with HCN, review the UC-Berkeley EH&S publication Toxic Gas Program on the EH&S website. If you have questions about Toxic Gas Program requirements, contact EH&S at 642-3073. 3. Potential Hazards/Toxicity Rev. Date: 09Sept2016 1 Hydrogen Cyanide Chemical Specific Standard Operating Procedure GHS Classification Flammable liquids - Category 1 Acute Oral Toxicity - Category 1 Acute Dermal Toxicity - Category 1 Acute Inhalation Toxicity - Category 1 Specific target organ toxicity (single exposure) - Category 1 Acute Aquatic Toxicity - Category 1 Chronic Aquatic Toxicity - Category 1 GHS Label elements, including precautionary statements SIGNAL WORD: DANGER Hazard Statement(s): H224 Extremely flammable liquid and vapour.