Application of Committee Neural Networks for Gene Expression
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Silencing of Phosphoinositide-Specific
ANTICANCER RESEARCH 34: 4069-4076 (2014) Silencing of Phosphoinositide-specific Phospholipase C ε Remodulates the Expression of the Phosphoinositide Signal Transduction Pathway in Human Osteosarcoma Cell Lines VINCENZA RITA LO VASCO1, MARTINA LEOPIZZI2, DANIELA STOPPOLONI3 and CARLO DELLA ROCCA2 Departments of 1Sense Organs , 2Medicine and Surgery Sciences and Biotechnologies and 3Biochemistry Sciences “A. Rossi Fanelli”, Sapienza University, Rome, Italy Abstract. Background: Ezrin, a member of the signal transduction pathway (5). The reduction of PIP2 ezrin–radixin–moesin family, is involved in the metastatic induces ezrin dissociation from the plasma membrane (6). spread of osteosarcoma. Ezrin binds phosphatydil inositol-4,5- The levels of PIP2 are regulated by the PI-specific bisphosphate (PIP2), a crucial molecule of the phospholipase C (PI-PLC) family (7), constituting thirteen phosphoinositide signal transduction pathway. PIP2 levels are enzymes divided into six sub-families on the basis of amino regulated by phosphoinositide-specific phospholipase C (PI- acid sequence, domain structure, mechanism of recruitment PLC) enzymes. PI-PLCε isoform, a well-characterized direct and tissue distribution (7-15). PI-PLCε, a direct effector of effector of rat sarcoma (RAS), is at a unique convergence RAS (14-15), might be the point of convergence for the point for the broad range of signaling pathways that promote broad range of signalling pathways that promote the RAS GTPase-mediated signalling. Materials and Methods. By RASGTPase-mediated signalling (16). using molecular biology methods and microscopic analyses, In previous studies, we suggested a relationship between we analyzed the expression of ezrin and PLC genes after PI-PLC expression and ezrin (17-18). -

Survival-Associated Metabolic Genes in Colon and Rectal Cancers
Survival-associated Metabolic Genes in Colon and Rectal Cancers Yanfen Cui ( [email protected] ) Tianjin Cancer Institute: Tianjin Tumor Hospital https://orcid.org/0000-0001-7760-7503 Baoai Han tianjin tumor hospital He Zhang tianjin tumor hospital Zhiyong Wang tianjin tumor hospital Hui Liu tianjin tumor hospital Fei Zhang tianjin tumor hospital Ruifang Niu tianjin tumor hospital Research Keywords: colon cancer, rectal cancer, prognosis, metabolism Posted Date: December 4th, 2020 DOI: https://doi.org/10.21203/rs.3.rs-117478/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 1/42 Abstract Background Uncontrolled proliferation is the most prominent biological feature of tumors. To rapidly proliferate and maximize the use of available nutrients, tumor cells regulate their metabolic behavior and the expression of metabolism-related genes (MRGs). In this study, we aimed to construct prognosis models for colon and rectal cancers, using MRGs to indicate the prognoses of patients. Methods We rst acquired the gene expression proles of colon and rectal cancers from the TCGA and GEO database, and utilized univariate Cox analysis, lasso regression, and multivariable cox analysis to identify MRGs for risk models. Then GSEA and KEGG functional enrichment analysis were utilized to identify the metabolism pathway of MRGs in the risk models and analyzed these genes comprehensively using GSCALite. Results Eight genes (CPT1C, PLCB2, PLA2G2D, GAMT, ENPP2, PIP4K2B, GPX3, and GSR) in the colon cancer risk model and six genes (TDO2, PKLR, GAMT, EARS2, ACO1, and WAS) in the rectal cancer risk model were identied successfully. Multivariate Cox analysis indicated that the models predicted overall survival accurately and independently for patients with colon or rectal cancer. -
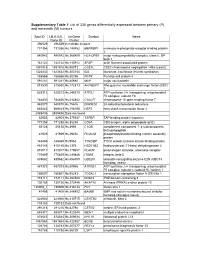
Supplementary Table 1 List of 335 Genes Differentially Expressed Between Primary (P) and Metastatic (M) Tumours
Supplementary Table 1 List of 335 genes differentially expressed between primary (P) and metastatic (M) tumours Spot ID I.M.A.G.E. UniGene Symbol Name Clone ID Cluster 296529 296529 In multiple clusters 731356 731356 Hs.140452 M6PRBP1 mannose-6-phosphate receptor binding protein 1 840942 840942 Hs.368409 HLA-DPB1 major histocompatibility complex, class II, DP beta 1 142122 142122 Hs.115912 AFAP actin filament associated protein 1891918 1891918 Hs.90073 CSE1L CSE1 chromosome segregation 1-like (yeast) 1323432 1323432 Hs.303154 IDS iduronate 2-sulfatase (Hunter syndrome) 788566 788566 Hs.80296 PCP4 Purkinje cell protein 4 591281 591281 Hs.80680 MVP major vault protein 815530 815530 Hs.172813 ARHGEF7 Rho guanine nucleotide exchange factor (GEF) 7 825312 825312 Hs.246310 ATP5J ATP synthase, H+ transporting, mitochondrial F0 complex, subunit F6 784830 784830 Hs.412842 C10orf7 chromosome 10 open reading frame 7 840878 840878 Hs.75616 DHCR24 24-dehydrocholesterol reductase 669443 669443 Hs.158195 HSF2 heat shock transcription factor 2 2485436 2485436 Data not found 82903 82903 Hs.370937 TAPBP TAP binding protein (tapasin) 771258 771258 Hs.85258 CD8A CD8 antigen, alpha polypeptide (p32) 85128 85128 Hs.8986 C1QB complement component 1, q subcomponent, beta polypeptide 41929 41929 Hs.39252 PICALM phosphatidylinositol binding clathrin assembly protein 148469 148469 Hs.9963 TYROBP TYRO protein tyrosine kinase binding protein 415145 415145 Hs.1376 HSD11B2 hydroxysteroid (11-beta) dehydrogenase 2 810017 810017 Hs.179657 PLAUR plasminogen activator, -

Supplementary Table S2
1-high in cerebrotropic Gene P-value patients Definition BCHE 2.00E-04 1 Butyrylcholinesterase PLCB2 2.00E-04 -1 Phospholipase C, beta 2 SF3B1 2.00E-04 -1 Splicing factor 3b, subunit 1 BCHE 0.00022 1 Butyrylcholinesterase ZNF721 0.00028 -1 Zinc finger protein 721 GNAI1 0.00044 1 Guanine nucleotide binding protein (G protein), alpha inhibiting activity polypeptide 1 GNAI1 0.00049 1 Guanine nucleotide binding protein (G protein), alpha inhibiting activity polypeptide 1 PDE1B 0.00069 -1 Phosphodiesterase 1B, calmodulin-dependent MCOLN2 0.00085 -1 Mucolipin 2 PGCP 0.00116 1 Plasma glutamate carboxypeptidase TMX4 0.00116 1 Thioredoxin-related transmembrane protein 4 C10orf11 0.00142 1 Chromosome 10 open reading frame 11 TRIM14 0.00156 -1 Tripartite motif-containing 14 APOBEC3D 0.00173 -1 Apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like 3D ANXA6 0.00185 -1 Annexin A6 NOS3 0.00209 -1 Nitric oxide synthase 3 SELI 0.00209 -1 Selenoprotein I NYNRIN 0.0023 -1 NYN domain and retroviral integrase containing ANKFY1 0.00253 -1 Ankyrin repeat and FYVE domain containing 1 APOBEC3F 0.00278 -1 Apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like 3F EBI2 0.00278 -1 Epstein-Barr virus induced gene 2 ETHE1 0.00278 1 Ethylmalonic encephalopathy 1 PDE7A 0.00278 -1 Phosphodiesterase 7A HLA-DOA 0.00305 -1 Major histocompatibility complex, class II, DO alpha SOX13 0.00305 1 SRY (sex determining region Y)-box 13 ABHD2 3.34E-03 1 Abhydrolase domain containing 2 MOCS2 0.00334 1 Molybdenum cofactor synthesis 2 TTLL6 0.00365 -1 Tubulin tyrosine ligase-like family, member 6 SHANK3 0.00394 -1 SH3 and multiple ankyrin repeat domains 3 ADCY4 0.004 -1 Adenylate cyclase 4 CD3D 0.004 -1 CD3d molecule, delta (CD3-TCR complex) (CD3D), transcript variant 1, mRNA. -

140503 IPF Signatures Supplement Withfigs Thorax
Supplementary material for Heterogeneous gene expression signatures correspond to distinct lung pathologies and biomarkers of disease severity in idiopathic pulmonary fibrosis Daryle J. DePianto1*, Sanjay Chandriani1⌘*, Alexander R. Abbas1, Guiquan Jia1, Elsa N. N’Diaye1, Patrick Caplazi1, Steven E. Kauder1, Sabyasachi Biswas1, Satyajit K. Karnik1#, Connie Ha1, Zora Modrusan1, Michael A. Matthay2, Jasleen Kukreja3, Harold R. Collard2, Jackson G. Egen1, Paul J. Wolters2§, and Joseph R. Arron1§ 1Genentech Research and Early Development, South San Francisco, CA 2Department of Medicine, University of California, San Francisco, CA 3Department of Surgery, University of California, San Francisco, CA ⌘Current address: Novartis Institutes for Biomedical Research, Emeryville, CA. #Current address: Gilead Sciences, Foster City, CA. *DJD and SC contributed equally to this manuscript §PJW and JRA co-directed this project Address correspondence to Paul J. Wolters, MD University of California, San Francisco Department of Medicine Box 0111 San Francisco, CA 94143-0111 [email protected] or Joseph R. Arron, MD, PhD Genentech, Inc. MS 231C 1 DNA Way South San Francisco, CA 94080 [email protected] 1 METHODS Human lung tissue samples Tissues were obtained at UCSF from clinical samples from IPF patients at the time of biopsy or lung transplantation. All patients were seen at UCSF and the diagnosis of IPF was established through multidisciplinary review of clinical, radiological, and pathological data according to criteria established by the consensus classification of the American Thoracic Society (ATS) and European Respiratory Society (ERS), Japanese Respiratory Society (JRS), and the Latin American Thoracic Association (ALAT) (ref. 5 in main text). Non-diseased normal lung tissues were procured from lungs not used by the Northern California Transplant Donor Network. -

Supp Table 6.Pdf
Supplementary Table 6. Processes associated to the 2037 SCL candidate target genes ID Symbol Entrez Gene Name Process NM_178114 AMIGO2 adhesion molecule with Ig-like domain 2 adhesion NM_033474 ARVCF armadillo repeat gene deletes in velocardiofacial syndrome adhesion NM_027060 BTBD9 BTB (POZ) domain containing 9 adhesion NM_001039149 CD226 CD226 molecule adhesion NM_010581 CD47 CD47 molecule adhesion NM_023370 CDH23 cadherin-like 23 adhesion NM_207298 CERCAM cerebral endothelial cell adhesion molecule adhesion NM_021719 CLDN15 claudin 15 adhesion NM_009902 CLDN3 claudin 3 adhesion NM_008779 CNTN3 contactin 3 (plasmacytoma associated) adhesion NM_015734 COL5A1 collagen, type V, alpha 1 adhesion NM_007803 CTTN cortactin adhesion NM_009142 CX3CL1 chemokine (C-X3-C motif) ligand 1 adhesion NM_031174 DSCAM Down syndrome cell adhesion molecule adhesion NM_145158 EMILIN2 elastin microfibril interfacer 2 adhesion NM_001081286 FAT1 FAT tumor suppressor homolog 1 (Drosophila) adhesion NM_001080814 FAT3 FAT tumor suppressor homolog 3 (Drosophila) adhesion NM_153795 FERMT3 fermitin family homolog 3 (Drosophila) adhesion NM_010494 ICAM2 intercellular adhesion molecule 2 adhesion NM_023892 ICAM4 (includes EG:3386) intercellular adhesion molecule 4 (Landsteiner-Wiener blood group)adhesion NM_001001979 MEGF10 multiple EGF-like-domains 10 adhesion NM_172522 MEGF11 multiple EGF-like-domains 11 adhesion NM_010739 MUC13 mucin 13, cell surface associated adhesion NM_013610 NINJ1 ninjurin 1 adhesion NM_016718 NINJ2 ninjurin 2 adhesion NM_172932 NLGN3 neuroligin -

Chemical Agent and Antibodies B-Raf Inhibitor RAF265
Supplemental Materials and Methods: Chemical agent and antibodies B-Raf inhibitor RAF265 [5-(2-(5-(trifluromethyl)-1H-imidazol-2-yl)pyridin-4-yloxy)-N-(4-trifluoromethyl)phenyl-1-methyl-1H-benzp{D, }imidazol-2- amine] was kindly provided by Novartis Pharma AG and dissolved in solvent ethanol:propylene glycol:2.5% tween-80 (percentage 6:23:71) for oral delivery to mice by gavage. Antibodies to phospho-ERK1/2 Thr202/Tyr204(4370), phosphoMEK1/2(2338 and 9121)), phospho-cyclin D1(3300), cyclin D1 (2978), PLK1 (4513) BIM (2933), BAX (2772), BCL2 (2876) were from Cell Signaling Technology. Additional antibodies for phospho-ERK1,2 detection for western blot were from Promega (V803A), and Santa Cruz (E-Y, SC7383). Total ERK antibody for western blot analysis was K-23 from Santa Cruz (SC-94). Ki67 antibody (ab833) was from ABCAM, Mcl1 antibody (559027) was from BD Biosciences, Factor VIII antibody was from Dako (A082), CD31 antibody was from Dianova, (DIA310), and Cot antibody was from Santa Cruz Biotechnology (sc-373677). For the cyclin D1 second antibody staining was with an Alexa Fluor 568 donkey anti-rabbit IgG (Invitrogen, A10042) (1:200 dilution). The pMEK1 fluorescence was developed using the Alexa Fluor 488 chicken anti-rabbit IgG second antibody (1:200 dilution).TUNEL staining kits were from Promega (G2350). Mouse Implant Studies: Biopsy tissues were delivered to research laboratory in ice-cold Dulbecco's Modified Eagle Medium (DMEM) buffer solution. As the tissue mass available from each biopsy was limited, we first passaged the biopsy tissue in Balb/c nu/Foxn1 athymic nude mice (6-8 weeks of age and weighing 22-25g, purchased from Harlan Sprague Dawley, USA) to increase the volume of tumor for further implantation. -

Small Molecule Protein–Protein Interaction Inhibitors As CNS Therapeutic Agents: Current Progress and Future Hurdles
Neuropsychopharmacology REVIEWS (2009) 34, 126–141 & 2009 Nature Publishing Group All rights reserved 0893-133X/09 $30.00 ............................................................................................................................................................... REVIEW 126 www.neuropsychopharmacology.org Small Molecule Protein–Protein Interaction Inhibitors as CNS Therapeutic Agents: Current Progress and Future Hurdles 1 ,1,2,3 Levi L Blazer and Richard R Neubig* 1Department of Pharmacology, University of Michigan Medical School, Ann Arbor, MI, USA; 2Department of Internal Medicine, University of Michigan Medical School, Ann Arbor, MI, USA; 3Center for Chemical Genomics, University of Michigan Medical School, Ann Arbor, MI, USA Protein–protein interactions are a crucial element in cellular function. The wealth of information currently available on intracellular-signaling pathways has led many to appreciate the untapped pool of potential drug targets that reside downstream of the commonly targeted receptors. Over the last two decades, there has been significant interest in developing therapeutics and chemical probes that inhibit specific protein–protein interactions. Although it has been a challenge to develop small molecules that are capable of occluding the large, often relatively featureless protein–protein interaction interface, there are increasing numbers of examples of small molecules that function in this manner with reasonable potency. This article will highlight the current progress in the development of small -

The Long-Chain Fatty Acid Receptors FFA1 and FFA4 Are Involved in Food
© 2020. Published by The Company of Biologists Ltd | Journal of Experimental Biology (2020) 223, jeb227330. doi:10.1242/jeb.227330 RESEARCH ARTICLE The long-chain fatty acid receptors FFA1 and FFA4 are involved in food intake regulation in fish brain Cristina Velasco, Marta Conde-Sieira, Sara Comesaña, Mauro Chivite, AdriánDıaz-Rú ́a, JesúsM.Mıgueź and JoséL. Soengas* ABSTRACT demonstrated that these FFARs are present in enteroendocrine cells We hypothesized that the free fatty acid receptors FFA1 and FFA4 might of the gastrointestinal tract (GIT) where they relate the detection of be involved in the anorectic response observed in fish after rising levels changes in LCFA to the synthesis and release of gastrointestinal of long-chain fatty acids (LCFAs) such as oleate. In one experiment we hormones (Lu et al., 2018). FFA1 and FFA4 are expressed not only in demonstrated that intracerebroventricular (i.c.v.) treatment of rainbow GIT, but also in a number of other tissues including liver, adipose trout with FFA1 and FFA4 agonists elicited an anorectic response 2, 6 tissue, taste buds and brain (Dragano et al., 2017; Kimura et al., and 24 h after treatment. In a second experiment, the same i.c.v. 2020). In brain regions like the hypothalamus and hindbrain, the treatment resulted after 2 h in an enhancement in the mRNA abundance presence of these receptors has been related to their putative role as of anorexigenic neuropeptides pomca1 and cartpt and a decrease in the fatty acid sensors involved in the regulation of food intake and energy values of orexigenic peptides npy and agrp1. -

Ncounter® Human Autoimmune Profiling Panel
nCounter® Human AutoImmune Profiling Panel - Gene and Probe Details Official Symbol Accession Alias / Previous Symbol Official Full Name Other targets or Isoform Information ACE NM_000789.2 DCP1;angiotensin I converting enzyme (peptidyl-dipeptidase A) 1 angiotensin I converting enzyme ACIN1 NM_001164815.1 ACINUS;apoptotic chromatin condensation inducer in the nucleus apoptotic chromatin condensation inducer 1 ACP5 NM_001611.3 acid phosphatase 5, tartrate resistant CTRN2;ARP1 (actin-related protein 1, yeast) homolog B (centractin beta),ARP1 actin-related ACTR1B NM_005735.3 protein 1 homolog B, centractin beta ARP1 actin related protein 1 homolog B ADAM17 NM_003183.4 TACE;tumor necrosis factor, alpha, converting enzyme ADAM metallopeptidase domain 17 ADAR NM_001111.3 IFI4,G1P1;interferon-induced protein 4 adenosine deaminase, RNA specific ADORA2A NM_000675.3 ADORA2 adenosine A2a receptor AGER NM_001136.3 advanced glycosylation end-product specific receptor AGT NM_000029.3 SERPINA8;serpin peptidase inhibitor, clade A, member 8 angiotensinogen AHR NM_001621.3 aryl hydrocarbon receptor AICDA NM_020661.2 activation-induced cytidine deaminase activation induced cytidine deaminase AIM2 NM_004833.1 absent in melanoma 2 APECED;autoimmune regulator (autoimmune polyendocrinopathy candidiasis ectodermal AIRE NM_000383.2 dystrophy) autoimmune regulator AKT1 NM_001014432.1 v-akt murine thymoma viral oncogene homolog 1 AKT serine/threonine kinase 1 AKT2 NM_001626.4 v-akt murine thymoma viral oncogene homolog 2 AKT serine/threonine kinase 2 AKT3 NM_005465.4 -

Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name phospholipase C, beta 2 Gene Symbol Plcb2 Organism Mouse Gene Summary Description Not Available Gene Aliases AI550384, B230205M18Rik, B230399N12 RefSeq Accession No. NC_000068.7, NT_039207.8 UniGene ID Mm.215156 Ensembl Gene ID ENSMUSG00000040061 Entrez Gene ID 18796 Assay Information Unique Assay ID qMmuCEP0057116 Assay Type Probe - Validation information is for the primer pair using SYBR® Green detection Detected Coding Transcript(s) ENSMUST00000102524, ENSMUST00000159756 Amplicon Context Sequence TCCAGACAGGATTGATGGAGTTAGCAGTAGGTGACAGCTTGGTTCGATACCGCC TCTTGGGGTCCCCTGGGAGGCCAAACAGTTCCACTTCCACATAGGTGCGTACAC TGCGCTCTGACAGGAACTGCCCAGAGA Amplicon Length (bp) 105 Chromosome Location 2:118713761-118713895 Assay Design Exonic Purification Desalted Validation Results Efficiency (%) 102 R2 0.9991 cDNA Cq 25.19 cDNA Tm (Celsius) 84.5 gDNA Cq 25.28 Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 1/4 PrimePCR™Assay Validation Report Plcb2, Mouse Amplification Plot Amplification of cDNA generated from 25 ng of universal reference RNA Melt Peak Melt curve analysis of above amplification Standard Curve Standard curve generated using 20 million copies of template diluted 10-fold to 20 copies Page 2/4 PrimePCR™Assay Validation Report Products used to generate validation data Real-Time PCR Instrument CFX384 Real-Time PCR Detection System Reverse Transcription Reagent iScript™ Advanced cDNA Synthesis Kit for RT-qPCR Real-Time PCR Supermix SsoAdvanced™ SYBR® Green Supermix Experimental Sample qPCR Mouse Reference Total RNA Data Interpretation Unique Assay ID This is a unique identifier that can be used to identify the assay in the literature and online. Detected Coding Transcript(s) This is a list of the Ensembl transcript ID(s) that this assay will detect. -

Morphometric, Hemodynamic, and Multi-Omics Analyses in Heart Failure Rats with Preserved Ejection Fraction
International Journal of Molecular Sciences Article Morphometric, Hemodynamic, and Multi-Omics Analyses in Heart Failure Rats with Preserved Ejection Fraction Wenxi Zhang 1, Huan Zhang 2, Weijuan Yao 2, Li Li 1, Pei Niu 1, Yunlong Huo 3,* and Wenchang Tan 1,4,5,* 1 Department of Mechanics and Engineering Science, College of Engineering, Peking University, Beijing 100871, China; [email protected] (W.Z.); [email protected] (L.L.); [email protected] (P.N.) 2 Hemorheology Center, Department of Physiology and Pathophysiology, School of Basic Medical Sciences, Peking University Health Science Center, Beijing 100191, China; [email protected] (H.Z.); [email protected] (W.Y.) 3 Institute of Mechanobiology & Medical Engineering, School of Life Sciences & Biotechnology, Shanghai Jiao Tong University, Shanghai 200240, China 4 PKU-HKUST Shenzhen-Hongkong Institution, Shenzhen 518057, China 5 Shenzhen Graduate School, Peking University, Shenzhen 518055, China * Correspondence: [email protected] (Y.H.); [email protected] (W.T.) Received: 13 January 2020; Accepted: 7 May 2020; Published: 9 May 2020 Abstract: (1) Background: There are no successive treatments for heart failure with preserved ejection fraction (HFpEF) because of complex interactions between environmental, histological, and genetic risk factors. The objective of the study is to investigate changes in cardiomyocytes and molecular networks associated with HFpEF. (2) Methods: Dahl salt-sensitive (DSS) rats developed HFpEF when fed with a high-salt (HS) diet for 7 weeks, which was confirmed by in vivo and ex vivo measurements. Shotgun proteomics, microarray, Western blot, and quantitative RT-PCR analyses were further carried out to investigate cellular and molecular mechanisms.