Predicting the Structure and Selectivity of Coiled-Coil Proteins
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

POLYMER STRUCTURE and CHARACTERIZATION Professor
POLYMER STRUCTURE AND CHARACTERIZATION Professor John A. Nairn Fall 2007 TABLE OF CONTENTS 1 INTRODUCTION 1 1.1 Definitions of Terms . 2 1.2 Course Goals . 5 2 POLYMER MOLECULAR WEIGHT 7 2.1 Introduction . 7 2.2 Number Average Molecular Weight . 9 2.3 Weight Average Molecular Weight . 10 2.4 Other Average Molecular Weights . 10 2.5 A Distribution of Molecular Weights . 11 2.6 Most Probable Molecular Weight Distribution . 12 3 MOLECULAR CONFORMATIONS 21 3.1 Introduction . 21 3.2 Nomenclature . 23 3.3 Property Calculation . 25 3.4 Freely-Jointed Chain . 27 3.4.1 Freely-Jointed Chain Analysis . 28 3.4.2 Comment on Freely-Jointed Chain . 34 3.5 Equivalent Freely Jointed Chain . 37 3.6 Vector Analysis of Polymer Conformations . 38 3.7 Freely-Rotating Chain . 41 3.8 Hindered Rotating Chain . 43 3.9 More Realistic Analysis . 45 3.10 Theta (Θ) Temperature . 47 3.11 Rotational Isomeric State Model . 48 4 RUBBER ELASTICITY 57 4.1 Introduction . 57 4.2 Historical Observations . 57 4.3 Thermodynamics . 60 4.4 Mechanical Properties . 62 4.5 Making Elastomers . 68 4.5.1 Diene Elastomers . 68 0 4.5.2 Nondiene Elastomers . 69 4.5.3 Thermoplastic Elastomers . 70 5 AMORPHOUS POLYMERS 73 5.1 Introduction . 73 5.2 The Glass Transition . 73 5.3 Free Volume Theory . 73 5.4 Physical Aging . 73 6 SEMICRYSTALLINE POLYMERS 75 6.1 Introduction . 75 6.2 Degree of Crystallization . 75 6.3 Structures . 75 Chapter 1 INTRODUCTION The topic of polymer structure and characterization covers molecular structure of polymer molecules, the arrangement of polymer molecules within a bulk polymer material, and techniques used to give information about structure or properties of polymers. -

Structural Biology of Laminins
Essays in Biochemistry (2019) EBC20180075 https://doi.org/10.1042/EBC20180075 Review Article Structural biology of laminins Erhard Hohenester Department of Life Sciences, Imperial College London, London SW7 2AZ, U.K. Correspondence: Erhard Hohenester ([email protected]) Laminins are large cell-adhesive glycoproteins that are required for the formation and func- tion of basement membranes in all animals. Structural studies by electron microscopy in the early 1980s revealed a cross-shaped molecule, which subsequently was shown to con- sist of three distinct polypeptide chains. Crystallographic studies since the mid-1990s have added atomic detail to all parts of the laminin heterotrimer. The three short arms of the cross are made up of continuous arrays of disulphide-rich domains. The globular domains at the tips of the short arms mediate laminin polymerization; the surface regions involved in this process have been identified by structure-based mutagenesis. The long arm of the cross is an α-helical coiled coil of all three chains, terminating in a cell-adhesive globular region. The molecular basis of cell adhesion to laminins has been revealed by recent structures of heterotrimeric integrin-binding fragments and of a laminin fragment bound to the carbohy- drate modification of dystroglycan. The structural characterization of the laminin molecule is essentially complete, but we still have to find ways of imaging native laminin polymers at molecular resolution. Introduction About 40 years ago, two laboratories independently purified a large glycoprotein from the extracellular matrix produced by mouse tumour cells [1,2]. Antibodies raised against this glycoprotein reacted with basement membranes (also known as basal laminae) in mouse tissues, prompting Rupert Timpl and col- leagues to name the new protein laminin. -

Ideal Chain Conformations and Statistics
ENAS 606 : Polymer Physics Chinedum Osuji 01.24.2013:HO2 Ideal Chain Conformations and Statistics 1 Overview Consideration of the structure of macromolecules starts with a look at the details of chain level chemical details which can impact the conformations adopted by the polymer. In the case of saturated carbon ◦ backbones, while maintaining the desired Ci−1 − Ci − Ci+1 bond angle of 112 , the placement of the final carbon in the triad above can occur at any point along the circumference of a circle, defining a torsion angle '. We can readily recognize the energetic differences as a function this angle, U(') such that there are 3 minima - a deep minimum corresponding the the trans state, for which ' = 0 and energetically equivalent gauche− and gauche+ states at ' = ±120 degrees, as shown in Fig 1. Figure 1: Trans, gauche- and gauche+ configurations, and their energetic sates 1.1 Static Flexibility The static flexibility of the chain in equilibrium is determined by the difference between the levels of the energy minima corresponding the gauche and trans states, ∆. If ∆ < kT , the g+, g− and t states occur with similar probability, and so the chain can change direction and appears as a random coil. If ∆ takes on a larger value, then the t conformations will be enriched, so the chain will be rigid locally, but on larger length scales, the eventual occurrence of g+ and g− conformations imparts a random conformation. Overall, if we ignore details on some length scale smaller than lp, the persistence length, the polymer appears as a continuous flexible chain where 1 lp = l0 exp(∆/kT ) (1) where l0 is something like a monomer length. -

Native-Like Mean Structure in the Unfolded Ensemble of Small Proteins
B doi:10.1016/S0022-2836(02)00888-4 available online at http://www.idealibrary.com on w J. Mol. Biol. (2002) 323, 153–164 Native-like Mean Structure in the Unfolded Ensemble of Small Proteins Bojan Zagrovic1, Christopher D. Snow1, Siraj Khaliq2 Michael R. Shirts2 and Vijay S. Pande1,2* 1Biophysics Program The nature of the unfolded state plays a great role in our understanding of Stanford University, Stanford proteins. However, accurately studying the unfolded state with computer CA 94305-5080, USA simulation is difficult, due to its complexity and the great deal of sampling required. Using a supercluster of over 10,000 processors we 2Department of Chemistry have performed close to 800 ms of molecular dynamics simulation in Stanford University, Stanford atomistic detail of the folded and unfolded states of three polypeptides CA 94305-5080, USA from a range of structural classes: the all-alpha villin headpiece molecule, the beta hairpin tryptophan zipper, and a designed alpha-beta zinc finger mimic. A comparison between the folded and the unfolded ensembles reveals that, even though virtually none of the individual members of the unfolded ensemble exhibits native-like features, the mean unfolded structure (averaged over the entire unfolded ensemble) has a native-like geometry. This suggests several novel implications for protein folding and structure prediction as well as new interpretations for experiments which find structure in ensemble-averaged measurements. q 2002 Elsevier Science Ltd. All rights reserved Keywords: mean-structure hypothesis; unfolded state of proteins; *Corresponding author distributed computing; conformational averaging Introduction under folding conditions, with some notable exceptions.14 – 16 This is understandable since under Historically, the unfolded state of proteins has such conditions the unfolded state is an unstable, received significantly less attention than the folded fleeting species making any kind of quantitative state.1 The reasons for this are primarily its struc- experimental measurement very difficult. -

Leucine Zippers
Leucine Zippers Leucine Zippers Advanced article Toshio Hakoshima, Nara Institute of Science and Technology, Nara, Japan Article contents Introduction The leucine zipper (ZIP) motif consists of a periodic repetition of a leucine residue at every Structural Basis of ZIP seventh position and forms an a-helical conformation, which facilitates dimerization and in Occurrence of ZIP and Coiled-coil Motifs some cases higher oligomerization of proteins. In many eukaryotic gene regulatory proteins, Dimerization Specificity of ZIP the ZIP motif is flanked at its N-terminus by a basic region containing characteristic residues DNA-binding Specificity of bZIP that facilitate DNA binding. doi: 10.1038/npg.els.0005049 Introduction protein modules for protein–protein interactions. Knowing the structure and function of these motifs A structure referred to as the leucine zipper or enables us to understand the molecular recognition simply as ZIP has been proposed to explain how a system in several biological processes. class of eukaryotic gene regulatory proteins works (Landschulz et al., 1988). A segment of the mammalian CCAAT/enhancer binding protein (C/EBP) of 30 Structural Basis of ZIP amino acids shares notable sequence similarity with a segment of the cellular Myc transforming protein. The The a helix is a secondary structure element that segments have been found to contain a periodic occurs frequently in proteins. Alpha helices are repetition of a leucine residue at every seventh stabilized in proteins by being packed into the position. A periodic array of at least four leucines hydrophobic core of a protein through hydrophobic has also been noted in the sequences of the Fos and side chains. -

Α/Β Coiled Coils 2 3 Marcus D
1 α/β Coiled Coils 2 3 Marcus D. Hartmann, Claudia T. Mendler†, Jens Bassler, Ioanna Karamichali, Oswin 4 Ridderbusch‡, Andrei N. Lupas* and Birte Hernandez Alvarez* 5 6 Department of Protein Evolution, Max Planck Institute for Developmental Biology, 72076 7 Tübingen, Germany 8 † present address: Nuklearmedizinische Klinik und Poliklinik, Klinikum rechts der Isar, 9 Technische Universität München, Munich, Germany 10 ‡ present address: Vossius & Partner, Siebertstraße 3, 81675 Munich, Germany 11 12 13 14 * correspondence to A. N. Lupas or B. Hernandez Alvarez: 15 Department of Protein Evolution 16 Max-Planck-Institute for Developmental Biology 17 Spemannstr. 35 18 D-72076 Tübingen 19 Germany 20 Tel. –49 7071 601 356 21 Fax –49 7071 601 349 22 [email protected], [email protected] 23 1 24 Abstract 25 Coiled coils are the best-understood protein fold, as their backbone structure can uniquely be 26 described by parametric equations. This level of understanding has allowed their manipulation 27 in unprecedented detail. They do not seem a likely source of surprises, yet we describe here 28 the unexpected formation of a new type of fiber by the simple insertion of two or six residues 29 into the underlying heptad repeat of a parallel, trimeric coiled coil. These insertions strain the 30 supercoil to the breaking point, causing the local formation of short β-strands, which move the 31 path of the chain by 120° around the trimer axis. The result is an α/β coiled coil, which retains 32 only one backbone hydrogen bond per repeat unit from the parent coiled coil. -
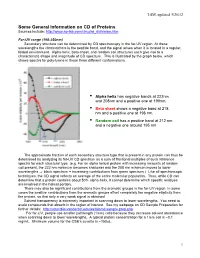
CD of Proteins Sources Include
LSM, updated 3/26/12 Some General Information on CD of Proteins Sources include: http://www.ap-lab.com/circular_dichroism.htm Far-UV range (190-250nm) Secondary structure can be determined by CD spectroscopy in the far-UV region. At these wavelengths the chromophore is the peptide bond, and the signal arises when it is located in a regular, folded environment. Alpha-helix, beta-sheet, and random coil structures each give rise to a characteristic shape and magnitude of CD spectrum. This is illustrated by the graph below, which shows spectra for poly-lysine in these three different conformations. • Alpha helix has negative bands at 222nm and 208nm and a positive one at 190nm. • Beta sheet shows a negative band at 218 nm and a positive one at 196 nm. • Random coil has a positive band at 212 nm and a negative one around 195 nm. The approximate fraction of each secondary structure type that is present in any protein can thus be determined by analyzing its far-UV CD spectrum as a sum of fractional multiples of such reference spectra for each structural type. (e.g. For an alpha helical protein with increasing amounts of random coil present, the 222 nm minimum becomes shallower and the 208 nm minimum moves to lower wavelengths ⇒ black spectrum + increasing contributions from green spectrum.) Like all spectroscopic techniques, the CD signal reflects an average of the entire molecular population. Thus, while CD can determine that a protein contains about 50% alpha-helix, it cannot determine which specific residues are involved in the helical portion. -

Folding and Unfolding of Helix-Turn-Helix Motifs in the Gas Phase
FOCUS:FROM MOBILITIES TO PROTEOMES Folding and Unfolding of Helix-Turn-Helix Motifs in the Gas Phase Lloyd W. Zilch, David T. Kaleta, Motoya Kohtani, Ranjani Krishnan, and Martin F. Jarrold Department of Chemistry, Indiana University, Bloomington, Indiana, USA Ion mobility measurements and molecular dynamic simulations have been performed for a series of peptides designed to have helix-turn-helix motifs. For peptides with two helical ϩ ϩ ϩ ϩ sections linked by a short loop region: AcA14KG3A14K 2H , AcA14KG5A14K 2H , ϩ ϩ ϩ ϩ ϭ ϭ ϭ AcA14KG7A14K 2H , and AcA14KSar3A14K 2H (Ac acetyl, A alanine, G glycine, Sar ϭ sarcosine and K ϭ lysine); a coiled-coil geometry with two anti-parallel helices is the lowest energy conformation. The helices uncouple and the coiled-coil unfolds as the temperature is raised. Equilibrium constants determined as a function of temperature yield enthalpy and entropy changes for the unfolding of the coiled-coil. The enthalpy and entropy changes depend on the length and nature of the loop region. For a peptide with three helical sections: protonated AcA14KG3A14KG3A14K; a coiled-coil bundle with three helices side-by-side is substantially less stable than a geometry with two helices in an antiparallel coiled-coil and the third helix collinear with one of the other two. (J Am Soc Mass Spectrom 2007, 18, 1239–1248) © 2007 American Society for Mass Spectrometry ϩ ϩ he function and activity of a protein is controlled AcA15K H helices are remarkably stable in the gas by its conformation. The conformation is deter- phase, and have been shown to remain intact to over ϩ ϩ Tmined by the protein’s potential energy surface, 700 K. -

Large-Scale Analyses of Site-Specific Evolutionary Rates Across
G C A T T A C G G C A T genes Article Large-Scale Analyses of Site-Specific Evolutionary Rates across Eukaryote Proteomes Reveal Confounding Interactions between Intrinsic Disorder, Secondary Structure, and Functional Domains Joseph B. Ahrens, Jordon Rahaman and Jessica Siltberg-Liberles * Department of Biological Sciences, Florida International University, Miami, FL 33199, USA; [email protected] (J.B.A.); jraha001@fiu.edu (J.R.) * Correspondence: jliberle@fiu.edu; Tel.: +1-305-348-7508 Received: 1 October 2018; Accepted: 9 November 2018; Published: 14 November 2018 Abstract: Various structural and functional constraints govern the evolution of protein sequences. As a result, the relative rates of amino acid replacement among sites within a protein can vary significantly. Previous large-scale work on Metazoan (Animal) protein sequence alignments indicated that amino acid replacement rates are partially driven by a complex interaction among three factors: intrinsic disorder propensity; secondary structure; and functional domain involvement. Here, we use sequence-based predictors to evaluate the effects of these factors on site-specific sequence evolutionary rates within four eukaryotic lineages: Metazoans; Plants; Saccharomycete Fungi; and Alveolate Protists. Our results show broad, consistent trends across all four Eukaryote groups. In all four lineages, there is a significant increase in amino acid replacement rates when comparing: (i) disordered vs. ordered sites; (ii) random coil sites vs. sites in secondary structures; and (iii) inter-domain linker sites vs. sites in functional domains. Additionally, within Metazoans, Plants, and Saccharomycetes, there is a strong confounding interaction between intrinsic disorder and secondary structure—alignment sites exhibiting both high disorder propensity and involvement in secondary structures have very low average rates of sequence evolution. -

Protein Folding and Structure Prediction
Protein Folding and Structure Prediction A Statistician's View Ingo Ruczinski Department of Biostatistics, Johns Hopkins University Proteins Amino acids without peptide bonds. Amino acids with peptide bonds. ¡ Amino acids are the building blocks of proteins. Proteins Both figures show the same protein (the bacterial protein L). The right figure also highlights the secondary structure elements. Space Resolution limit of a light microscope Glucose Ribosome Red blood cell C−C bond Hemoglobin Bacterium 1 10 100 1000 10000 100000 1nm 1µm Distance [ A° ] Energy C−C bond Green Noncovalent bond light Glucose Thermal ATP 0.1 1 10 100 1000 Energy [ kcal/mol ] Non-Bonding Interactions Amino acids of a protein are joined by covalent bonding interactions. The polypep- tide is folded in three dimension by non-bonding interactions. These interactions, which can easily be disrupted by extreme pH, temperature, pressure, and denatu- rants, are: Electrostatic Interactions (5 kcal/mol) Hydrogen-bond Interactions (3-7 kcal/mol) Van Der Waals Interactions (1 kcal/mol) ¡ Hydrophobic Interactions ( 10 kcal/mol) The total inter-atomic force acting between two atoms is the sum of all the forces they exert on each other. Energy Profile Transition State Denatured State Native State Radius of Gyration of Denatured Proteins Do chemically denatured proteins behave as random coils? The radius of gyration Rg of a protein is defined as the root mean square dis- tance from each atom of the protein to their centroid. For an ideal (infinitely thin) random-coil chain in a solvent, the average radius 0.5 ¡ of gyration of a random coil is a simple function of its length n: Rg n For an excluded volume polymer (a polymer with non-zero thickness and non- trivial interactions between monomers) in a solvent, the average radius of gyra- 0.588 tion, we have Rg n (Flory 1953). -

And Beta-Helical Protein Motifs
Soft Matter Mechanical Unfolding of Alpha- and Beta-helical Protein Motifs Journal: Soft Matter Manuscript ID SM-ART-10-2018-002046.R1 Article Type: Paper Date Submitted by the 28-Nov-2018 Author: Complete List of Authors: DeBenedictis, Elizabeth; Northwestern University Keten, Sinan; Northwestern University, Mechanical Engineering Page 1 of 10 Please doSoft not Matter adjust margins Soft Matter ARTICLE Mechanical Unfolding of Alpha- and Beta-helical Protein Motifs E. P. DeBenedictis and S. Keten* Received 24th September 2018, Alpha helices and beta sheets are the two most common secondary structure motifs in proteins. Beta-helical structures Accepted 00th January 20xx merge features of the two motifs, containing two or three beta-sheet faces connected by loops or turns in a single protein. Beta-helical structures form the basis of proteins with diverse mechanical functions such as bacterial adhesins, phage cell- DOI: 10.1039/x0xx00000x puncture devices, antifreeze proteins, and extracellular matrices. Alpha helices are commonly found in cellular and extracellular matrix components, whereas beta-helices such as curli fibrils are more common as bacterial and biofilm matrix www.rsc.org/ components. It is currently not known whether it may be advantageous to use one helical motif over the other for different structural and mechanical functions. To better understand the mechanical implications of using different helix motifs in networks, here we use Steered Molecular Dynamics (SMD) simulations to mechanically unfold multiple alpha- and beta- helical proteins at constant velocity at the single molecule scale. We focus on the energy dissipated during unfolding as a means of comparison between proteins and work normalized by protein characteristics (initial and final length, # H-bonds, # residues, etc.). -

Rapid and Accurate Prediction of Coiled-Coil Structures and Application to Modelling Intermediate filaments
bioRxiv preprint doi: https://doi.org/10.1101/123869; this version posted April 14, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. CCFold: rapid and accurate prediction of coiled-coil structures and application to modelling intermediate filaments Dmytro Guzenko and Sergei V. Strelkov ∗ Department of Pharmaceutical and Pharmacological Sciences KU Leuven, Leuven 3000, Belgium. ∗To whom correspondence should be addressed. Abstract Accurate molecular structure of the protein dimer representing the elementary building block of intermediate filaments (IFs) is essential towards the understanding of the filament assembly, rationalizing their mechanical properties and explaining the effect of disease- related IF mutations. The dimer contains a ∼300-residue long α-helical coiled coil which is not assessable to either direct experimental structure determination or modelling using standard approaches. At the same time, coiled coils are well-represented in structural databases. Here we present CCFold, a generally applicable threading-based algorithm which produces coiled-coil models from protein sequence only. The algorithm is based on a statistical analysis of experimentally determined structures and can handle any hydrophobic repeat patterns in addition to the most common heptads. We demonstrate that CCFold outperforms general-purpose computational folding in terms of accuracy, while being faster by orders of magnitude. By combining the CCFold algorithm and Rosetta folding we generate representative dimer models for all IF protein classes. The source code is freely available at https://github.com/biocryst/IF 1 Introduction Intermediate filaments (IFs) are an important example of a protein assembly based on α-helical coiled coils (CCs).