Dissertatio Final Copy
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
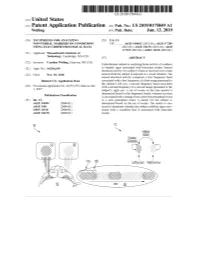
ANNNNNNNNNNNNNNNNNNNN 100A 006 Left Eye Input Right Eye Input
US 20190175049A1 ( 19) United States (12 ) Patent Application Publication (10 ) Pub. No. : US 2019 /0175049 A1 Welling ( 43 ) Pub . Date : Jun . 13 , 2019 ( 54 ) TECHNIQUES FOR ANALYZING (52 ) U . S . CI. NON -VERBAL MARKERS OF CONDITIONS CPC . .. A61B 5 /04842 (2013 . 01 ) ; A61B 5 / 7289 USING ELECTROPHYSIOLOGICAL DATA (2013 . 01) ; A61B 5 /0478 ( 2013 .01 ) ; A61B 5 /7225 ( 2013. 01 ) ; G06N 20 / 10 (2019 .01 ) (71 ) Applicant: Massachusetts Institute of Technology , Cambridge , MA (US ) ( 57 ) ABSTRACT (72 ) Inventor : Caroline Welling, Hanover, NH (US ) Embodiments related to analyzing brain activity of a subject to identify signs associated with binocular rivalry . Sensed ( 21 ) Appl. No. : 16 / 206, 639 electrical activity of a subject' s brain is received over a time period while the subject is exposed to a visual stimulus. The ( 22 ) Filed : Nov. 30 , 2018 sensed electrical activity comprises a first frequency band Related U . S . Application Data associated with a first frequency of a first image presented to the subject ' s left eye , a second frequency band associated (60 ) Provisional application No .62 / 593 , 535, filed on Dec . with a second frequency of a second image presented to the 1 , 2017 subject ' s right eye . A set of events in the time period is determined based on the frequency bands, wherein an event Publication Classification is associated with a change from a previous perceptual event (51 ) Int. Ci. to a new perceptual event. A metric for the subject is A61B 5 /0484 ( 2006 .01 ) determined based on the set of events . The metric is ana A61B 5 /00 ( 2006 .01 ) lyzed to determine whether the subject exhibits signs asso GO6N 20 / 10 (2006 .01 ) ciated with a condition that is associated with binocular A61B 5 /0478 ( 2006 .01 ) rivalry . -

(12) Patent Application Publication (10) Pub. No.: US 2006/0110428A1 De Juan Et Al
US 200601 10428A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2006/0110428A1 de Juan et al. (43) Pub. Date: May 25, 2006 (54) METHODS AND DEVICES FOR THE Publication Classification TREATMENT OF OCULAR CONDITIONS (51) Int. Cl. (76) Inventors: Eugene de Juan, LaCanada, CA (US); A6F 2/00 (2006.01) Signe E. Varner, Los Angeles, CA (52) U.S. Cl. .............................................................. 424/427 (US); Laurie R. Lawin, New Brighton, MN (US) (57) ABSTRACT Correspondence Address: Featured is a method for instilling one or more bioactive SCOTT PRIBNOW agents into ocular tissue within an eye of a patient for the Kagan Binder, PLLC treatment of an ocular condition, the method comprising Suite 200 concurrently using at least two of the following bioactive 221 Main Street North agent delivery methods (A)-(C): Stillwater, MN 55082 (US) (A) implanting a Sustained release delivery device com (21) Appl. No.: 11/175,850 prising one or more bioactive agents in a posterior region of the eye so that it delivers the one or more (22) Filed: Jul. 5, 2005 bioactive agents into the vitreous humor of the eye; (B) instilling (e.g., injecting or implanting) one or more Related U.S. Application Data bioactive agents Subretinally; and (60) Provisional application No. 60/585,236, filed on Jul. (C) instilling (e.g., injecting or delivering by ocular ion 2, 2004. Provisional application No. 60/669,701, filed tophoresis) one or more bioactive agents into the Vit on Apr. 8, 2005. reous humor of the eye. Patent Application Publication May 25, 2006 Sheet 1 of 22 US 2006/0110428A1 R 2 2 C.6 Fig. -

Calcium Channel Blocker As a Drug Candidate for the Treatment of Generalised Epilepsies
UNIVERSITAT DE BARCELONA Faculty of Pharmacy and Food Sciences Calcium channel blocker as a drug candidate for the treatment of generalised epilepsies Final degree project Author: Janire Sanz Sevilla Bachelor's degree in Pharmacy Primary field: Organic Chemistry, Pharmacology and Therapeutics Secondary field: Physiology, Pathophysiology and Molecular Biology March 2019 This work is licensed under a Creative Commons license ABBREVIATIONS AED antiepileptic drug AMPA α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid ANNA-1 antineuronal nuclear antibody 1 BBB blood-brain barrier Bn benzyl BnBr benzyl bromide BnNCO benzyl isocyanate Boc tert-butoxycarbonyl Bu4NBr tetrabutylammonium bromide Ca+2 calcium ion CACNA1 calcium channel voltage-dependent gene cAMP cyclic adenosine monophosphate CCB calcium channel blocker cGMP cyclic guanosine monophosphate CH3CN acetonitrile Cl- chlorine ion Cmax maximum concentration CMV cytomegalovirus CTScan computed axial tomography DCM dichloromethane DIPEA N,N-diisopropylethylamine DMF dimethylformamide DMPK drug metabolism and pharmacokinetics DNET dysembryoplastic neuroepithelial tumours EEG electroencephalogram EPSP excitatory post-synaptic potential FDA food and drug administration Fe iron FLIPR fluorescence imaging plate reader fMRI functional magnetic resonance imaging GABA γ-amino-α-hydroxybutyric acid GAD65 glutamic acid decarboxylase 65 GAERS generalised absence epilepsy rat of Strasbourg GluR5 kainate receptor GTC generalised tonic-clonic H+ hydrogen ion H2 hydrogen H2O dihydrogen dioxide (water) -

A Comparative Study of Progabide, Valproate, and Epilepsy
J Neurol Neurosurg Psychiatry: first published as 10.1136/jnnp.49.11.1251 on 1 November 1986. Downloaded from Journal of Neurology, Neurosurgery, and Psychiatry 1986;49:1251-1257 A comparative study of progabide, valproate, and placebo as add-on therapy in patients with refractory epilepsy P CRAWFORD, D CHADWICK From the Department ofNeurology, Walton Hospital, Liverpool, UK SUMMARY A three way single blind cross-over comparison of progabide, valproate and placebo, as adjunctive therapy, was undertaken in 64 patients with therapy-resistant partial and generalised seizures. The study was not completed because of the incidence of elevated hepatic enzymes on progabide. Analysis of efficacy showed progabide to be inferior to valproate against all seizure types, particularly against tonic-clonic seizures. Valproate was superior to placebo against all seizure types, partial and tonic-clonic seizures. Progabide did not differ significantly from placebo in any instance. In addition progabide caused elevation of hepatic enzymes which was symptomatic in one case, and was associated with an interaction with phenytoin symptoms which resulted in of guest. Protected by copyright. intoxication in some cases. Progabide is a pro-drug and a GABA agonist which treatment limb was of six months with a two week washout possesses anticonvulsant properties in a variety of and cross-over period between treatment phases. Patients experimental models of seizures and epilepsy.' with severe, partial or generalised epilepsies were eligible for Whether it possesses antiepileptic properties in man is admission to the study as long as they had a definite history controversial. A number of double blind of epilepsy confirmed by observation and EEG recording studies and suffered a minimum of one seizure per month during the against placebo have been reported, some of which six months prior to entry into the study. -

Late Shri Vishnu Waman Thakur Charitable Trust`S VIVA INSTITUTE of PHARMACY At: Shirgaon, Veer Sawarkar Road, Virar (E), Taluka: Vasai, Dist
Late Shri Vishnu Waman Thakur Charitable Trust`s VIVA INSTITUTE OF PHARMACY At: Shirgaon, Veer Sawarkar Road, Virar (E), Taluka: Vasai, Dist. Palghar-401305, Maharashtra. FINAL YEAR UNIVERSITY EXAMINATION 2019-2020 Final Year B.Pharm. Semester VIII SUBJECT-BPH_C_801_T-Pharmaceutical Chemistry III MULTIPLE CHOICE QUESTIONS: PRACTICE QUESTION BANK Q. 1 Which is the correct IUPAC name for the following structure? A] 5-chloro-2-(methylamino)-5-phenyl-3H-1,4-benzodiazepine B] 7-chloro-2-(methylamino)-5-pyridinyl-3H-1,4-benzodiazepine-4-oxide C] 7-chloro-2-(ethylamino)-5-phenyl-3H-1,5-benzodiazepine D] 7-chloro-2-(methylamino)-5-phenyl-3H-1,4-benzodiazepine-4-oxide Q. 2 Which of the following is long acting sedative hypnotic? A] Diazepam B] Alprazolam C] Temazepam D] Imipramine Q. 3 Name of oxide derivative used as sedative hypnotic is A] Diazepam B] Chlordiazepoxide C] Nitazepam D] Ramelteon Late Shri Vishnu Waman Thakur Charitable Trust`s VIVA INSTITUTE OF PHARMACY At: Shirgaon, Veer Sawarkar Road, Virar (E), Taluka: Vasai, Dist. Palghar-401305, Maharashtra. Q. 4 With respect to the following general structure which is the correct statement ? A] X must be electropositive substituent for optimum activity B] X must be aromatic ring for optimum activity C] X must be electronegative substituent for optimum activity D] X must be H for optimum activity Q. 5 Which is the incorrect statement with respect to structure given in Q. 4 A] Ring C is ortho substituted with electron withdrawing group for optimum activity B] Ring C when para substituted increases activity C] Ring C is diortho substituted with electron withdrawing group for optimum activity D] Ring C when para substituted decreases activity Q. -

Pharmaceutical Chemistry III MULTIPLE CHOICE QUESTIONS: PRACTICE QUESTION BANK
FINAL YEAR UNIVERSITY EXAMINATION 2019-2020 Final Year B.Pharm. Semester VIII SUBJECT-BPH_C_801_T-Pharmaceutical Chemistry III MULTIPLE CHOICE QUESTIONS: PRACTICE QUESTION BANK SET-I Q. 1 Which is the correct IUPAC name for the following structure? A] 5-chloro-2-(methylamino)-5-phenyl-3H-1,4-benzodiazepine B] 7-chloro-2-(methylamino)-5-pyridinyl-3H-1,4-benzodiazepine-4-oxide C] 7-chloro-2-(ethylamino)-5-phenyl-3H-1,5-benzodiazepine D] 7-chloro-2-(methylamino)-5-phenyl-3H-1,4-benzodiazepine-4-oxide Q. 2 Which of the following is long acting sedative hypnotic? A] Diazepam B] Alprazolam C] Temazepam D] Imipramine Q. 3 Name of oxide derivative used as sedative hypnotic is A] Diazepam B] Chlordiazepoxide C]Nitazepam D] Ramelteon Q. 4 With respect to the following general structure which is the correctstatement ? A] X must be electropositive substituent for optimum activity B] X must be aromatic ring for optimum activity C] X must be electronegative substituent for optimum activity D] X must be H for optimum activity Q. 5 Which is the incorrect statement with respect to structure given in Q. 4 A] Ring C is ortho substituted with electron withdrawing group for optimum activity B] Ring C when para substituted increases activity C] Ring C is diortho substituted with electron withdrawing group for optimum activity D] Ring C when para substituted decreases activity Q. 6 What is the starting material for synthesis of Piroxicam (structure given below) A] B] C] D] Q. 7 Which one of the following is Cytokine inhibitor? A] Abatacept B] Fluoxetine C] Propranolol D]Aldosterone Q. -

Pharmaceuticals As Environmental Contaminants
PharmaceuticalsPharmaceuticals asas EnvironmentalEnvironmental Contaminants:Contaminants: anan OverviewOverview ofof thethe ScienceScience Christian G. Daughton, Ph.D. Chief, Environmental Chemistry Branch Environmental Sciences Division National Exposure Research Laboratory Office of Research and Development Environmental Protection Agency Las Vegas, Nevada 89119 [email protected] Office of Research and Development National Exposure Research Laboratory, Environmental Sciences Division, Las Vegas, Nevada Why and how do drugs contaminate the environment? What might it all mean? How do we prevent it? Office of Research and Development National Exposure Research Laboratory, Environmental Sciences Division, Las Vegas, Nevada This talk presents only a cursory overview of some of the many science issues surrounding the topic of pharmaceuticals as environmental contaminants Office of Research and Development National Exposure Research Laboratory, Environmental Sciences Division, Las Vegas, Nevada A Clarification We sometimes loosely (but incorrectly) refer to drugs, medicines, medications, or pharmaceuticals as being the substances that contaminant the environment. The actual environmental contaminants, however, are the active pharmaceutical ingredients – APIs. These terms are all often used interchangeably Office of Research and Development National Exposure Research Laboratory, Environmental Sciences Division, Las Vegas, Nevada Office of Research and Development Available: http://www.epa.gov/nerlesd1/chemistry/pharma/image/drawing.pdfNational -

Marrakesh Agreement Establishing the World Trade Organization
No. 31874 Multilateral Marrakesh Agreement establishing the World Trade Organ ization (with final act, annexes and protocol). Concluded at Marrakesh on 15 April 1994 Authentic texts: English, French and Spanish. Registered by the Director-General of the World Trade Organization, acting on behalf of the Parties, on 1 June 1995. Multilat ral Accord de Marrakech instituant l©Organisation mondiale du commerce (avec acte final, annexes et protocole). Conclu Marrakech le 15 avril 1994 Textes authentiques : anglais, français et espagnol. Enregistré par le Directeur général de l'Organisation mondiale du com merce, agissant au nom des Parties, le 1er juin 1995. Vol. 1867, 1-31874 4_________United Nations — Treaty Series • Nations Unies — Recueil des Traités 1995 Table of contents Table des matières Indice [Volume 1867] FINAL ACT EMBODYING THE RESULTS OF THE URUGUAY ROUND OF MULTILATERAL TRADE NEGOTIATIONS ACTE FINAL REPRENANT LES RESULTATS DES NEGOCIATIONS COMMERCIALES MULTILATERALES DU CYCLE D©URUGUAY ACTA FINAL EN QUE SE INCORPOR N LOS RESULTADOS DE LA RONDA URUGUAY DE NEGOCIACIONES COMERCIALES MULTILATERALES SIGNATURES - SIGNATURES - FIRMAS MINISTERIAL DECISIONS, DECLARATIONS AND UNDERSTANDING DECISIONS, DECLARATIONS ET MEMORANDUM D©ACCORD MINISTERIELS DECISIONES, DECLARACIONES Y ENTEND MIENTO MINISTERIALES MARRAKESH AGREEMENT ESTABLISHING THE WORLD TRADE ORGANIZATION ACCORD DE MARRAKECH INSTITUANT L©ORGANISATION MONDIALE DU COMMERCE ACUERDO DE MARRAKECH POR EL QUE SE ESTABLECE LA ORGANIZACI N MUND1AL DEL COMERCIO ANNEX 1 ANNEXE 1 ANEXO 1 ANNEX -

BMJ Open Is Committed to Open Peer Review. As Part of This Commitment We Make the Peer Review History of Every Article We Publish Publicly Available
BMJ Open is committed to open peer review. As part of this commitment we make the peer review history of every article we publish publicly available. When an article is published we post the peer reviewers’ comments and the authors’ responses online. We also post the versions of the paper that were used during peer review. These are the versions that the peer review comments apply to. The versions of the paper that follow are the versions that were submitted during the peer review process. They are not the versions of record or the final published versions. They should not be cited or distributed as the published version of this manuscript. BMJ Open is an open access journal and the full, final, typeset and author-corrected version of record of the manuscript is available on our site with no access controls, subscription charges or pay-per-view fees (http://bmjopen.bmj.com). If you have any questions on BMJ Open’s open peer review process please email [email protected] BMJ Open Pediatric drug utilization in the Western Pacific region: Australia, Japan, South Korea, Hong Kong and Taiwan Journal: BMJ Open ManuscriptFor ID peerbmjopen-2019-032426 review only Article Type: Research Date Submitted by the 27-Jun-2019 Author: Complete List of Authors: Brauer, Ruth; University College London, Research Department of Practice and Policy, School of Pharmacy Wong, Ian; University College London, Research Department of Practice and Policy, School of Pharmacy; University of Hong Kong, Centre for Safe Medication Practice and Research, Department -

Anticonvulsant Effect of Progabide in Rats During Ontogenesis
Physiol. Res. 46: 47- 52, 1997 Anticonvulsant Effect of Progabide in Rats during Ontogenesis L. STAŇKOVÁ1, A. KOŽUCHOVÁ1, P. MAREŠ12 institute of Physiology, Academy of Sciences of the Czech Republic and 2Department of Pathophysiology, Third Faculty o f Medicine, Charles University, Prague, Czech Republic Received July 13, 1995 Accepted August 28, 1996 Summary The action of progabide against motor seizures elicited by pentylenetetrazol was studied in 7-, 12-, 18-, 25-day-old and adult rats. Progabide (dissolved in dimethylsulfoxide) was injected in doses from 12.5 to 150 mg/kg i.p. 30 min before pentylenetetrazol. Minimal seizures were not affected by solvent or progabide pretreatment. The action of progabide against major, i.e. generalized tonic-clonic seizures, changed with age: adult rats exhibited a tendency to suppression of whole major seizures, whereas specific suppression of the tonic phase was observed in rat pups during the first three weeks of life. The only effect seen in 25-day-old animals was prolongation of the latency of major seizures after the highest dose of progabide. Key words Motor seizures — Rat - Ontogenesis - Progabide - Pentylenetetrazol Introduction Progabide (PGB, 4-{[4-(chlorophenyl) (5- fluoro-2-hydroxyphenyl)methylene]amino} butanamide see Fig. 1) is an anticonvulsant drug introduced into clinical in the eighties (for review see Morselli et al. 1986, Morselli and Palminteri 1989, Loiseau and Duché 1990). Biochemical and pharmacological experiments Cl Cl have demonstrated that PGB as well as its metabolite SL 75102 (Fig. 1) exhibit GABAmimetic effects by direct stimulation of the GABA receptors (Lloyd et al. Progabide SL 75 102 1982, Langer et al. -

Federal Register / Vol. 60, No. 80 / Wednesday, April 26, 1995 / Notices DIX to the HTSUS—Continued
20558 Federal Register / Vol. 60, No. 80 / Wednesday, April 26, 1995 / Notices DEPARMENT OF THE TREASURY Services, U.S. Customs Service, 1301 TABLE 1.ÐPHARMACEUTICAL APPEN- Constitution Avenue NW, Washington, DIX TO THE HTSUSÐContinued Customs Service D.C. 20229 at (202) 927±1060. CAS No. Pharmaceutical [T.D. 95±33] Dated: April 14, 1995. 52±78±8 ..................... NORETHANDROLONE. A. W. Tennant, 52±86±8 ..................... HALOPERIDOL. Pharmaceutical Tables 1 and 3 of the Director, Office of Laboratories and Scientific 52±88±0 ..................... ATROPINE METHONITRATE. HTSUS 52±90±4 ..................... CYSTEINE. Services. 53±03±2 ..................... PREDNISONE. 53±06±5 ..................... CORTISONE. AGENCY: Customs Service, Department TABLE 1.ÐPHARMACEUTICAL 53±10±1 ..................... HYDROXYDIONE SODIUM SUCCI- of the Treasury. NATE. APPENDIX TO THE HTSUS 53±16±7 ..................... ESTRONE. ACTION: Listing of the products found in 53±18±9 ..................... BIETASERPINE. Table 1 and Table 3 of the CAS No. Pharmaceutical 53±19±0 ..................... MITOTANE. 53±31±6 ..................... MEDIBAZINE. Pharmaceutical Appendix to the N/A ............................. ACTAGARDIN. 53±33±8 ..................... PARAMETHASONE. Harmonized Tariff Schedule of the N/A ............................. ARDACIN. 53±34±9 ..................... FLUPREDNISOLONE. N/A ............................. BICIROMAB. 53±39±4 ..................... OXANDROLONE. United States of America in Chemical N/A ............................. CELUCLORAL. 53±43±0 -

Assessment Report: the Clinical Effectiveness and Cost-Effectiveness
The clinical effectiveness and cost-effectiveness of newer drugs for children with epilepsy Report commissioned by: NHS R&D HTA Programme On behalf of: The National Institute for Clinical Excellence Produced by: West Midlands Health Technology Assessment Collaboration, Department of Public Health and Epidemiology, The University of Birmingham Authors: alphabetical Prof Stirling Bryan Senior health economist# Dr Martin Connock Systematic reviewerà Dr Carole Cummins Paediatric trials adviser† Emma Frew Health economist# Anne Fry-Smith Information specialistà Beti-Wynn Jones Systematic reviewer‡ Prof Alain Li Wan Po Pharmacological adviser†† Josie Sandercock Senior reviewerà à Department of Public Health and Epidemiology, The University of Birmingham. # Health Economics Facility, Health Services Management Centre, The University of Birmingham. † Institute of Child Health, Whittal Street, Birmingham. †† Centre for Evidence-Based Pharmacotherapy, Aston University, Birmingham. ‡ Department of Medicine Management, Keele University Correspondence to: J. Sandercock, Tel 0121-414-2247. Email [email protected] M. Connock, Tel 0121-414-7507. Email [email protected] West Midlands Health Technology Collaboration, Department of Public Health and Epidemiology, The University of Birmingham, Edgbaston, Birmingham B15 2TT. FAX 0121- 414-7878 Date completed: February 2003 Expiry Date: Expiry date Newer Drugs for Children with Epilepsy PUBLICATION INFORMATION How to reference this publication: Connock M, Frew E, Jones B, Bryan S, Cummins C, Fry-Smith A, Li Wan Po A, Sandercock J. The clinical effectiveness and cost effectiveness of newer drugs for children with epilepsy. Birmingham: West Midlands Health Technology Collaboration, University of Birmingham, February 2003 ABOUT “HOME UNIT” The West Midlands Health Technology Assessment Collaboration (WMHTAC) produces rapid systematic reviews about the effectiveness of healthcare interventions and technologies, in response to requests from West Midlands Health Authorities or the HTA programme.