Prediction of Chemical Reactivity Parameters and Physical Properties of Organic Compounds from Molecular Structure Using Sparc
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Brief Guide to the Nomenclature of Organic Chemistry
1 Brief Guide to the Nomenclature of Table 1: Components of the substitutive name Organic Chemistry (4S,5E)-4,6-dichlorohept-5-en-2-one for K.-H. Hellwich (Germany), R. M. Hartshorn (New Zealand), CH3 Cl O A. Yerin (Russia), T. Damhus (Denmark), A. T. Hutton (South 4 2 Africa). E-mail: [email protected] Sponsoring body: Cl 6 CH 5 3 IUPAC Division of Chemical Nomenclature and Structure suffix for principal hept(a) parent (heptane) one Representation. characteristic group en(e) unsaturation ending chloro substituent prefix 1 INTRODUCTION di multiplicative prefix S E stereodescriptors CHEMISTRY The universal adoption of an agreed nomenclature is a key tool for 2 4 5 6 locants ( ) enclosing marks efficient communication in the chemical sciences, in industry and Multiplicative prefixes (Table 2) are used when more than one for regulations associated with import/export or health and safety. fragment of a particular kind is present in a structure. Which kind of REPRESENTATION The International Union of Pure and Applied Chemistry (IUPAC) multiplicative prefix is used depends on the complexity of the provides recommendations on many aspects of nomenclature.1 The APPLIED corresponding fragment – e.g. trichloro, but tris(chloromethyl). basics of organic nomenclature are summarized here, and there are companion documents on the nomenclature of inorganic2 and Table 2: Multiplicative prefixes for simple/complicated entities polymer3 chemistry, with hyperlinks to original documents. An No. Simple Complicated No. Simple Complicated AND overall -

Interstellar Formamide (NH2CHO), a Key Prebiotic Precursor
Interstellar formamide (NH2CHO), a key prebiotic precursor Ana López-Sepulcre,∗,y,z Nadia Balucani,{ Cecilia Ceccarelli,y Claudio Codella,x,y François Dulieu,k and Patrice Theulé? yUniv. Grenoble Alpes, IPAG, F-38000 Grenoble, France zInstitut de Radioastronomie Millimétrique (IRAM), 300 rue de la piscine, F-38406 Saint-Martin d’Hères, France {Dipartimento di Chimica, Biologia e Biotecnologie, Università degli Studi di Perugia, I-06123 Perugia, Italy xINAF, Osservatorio Astrofisico di Arcetri, Largo E. Fermi 5, I-50125, Firenze, Italy kUniversité de Cergy-Pontoise, Observatoire de Paris, PSL University, Sorbonne Université, CNRS, LERMA, F-95000, Cergy-Pontoise, France ?Aix-Marseille Université, PIIM UMR-CNRS 7345, 13397 Marseille, France E-mail: [email protected] Abstract Formamide (NH2CHO) has been identified as a potential precursor of a wide variety arXiv:1909.11770v1 [astro-ph.SR] 25 Sep 2019 of organic compounds essential to life, and many biochemical studies propose it likely played a crucial role in the context of the origin of life on our planet. The detection of formamide in comets, which are believed to have –at least partially– inherited their current chemical composition during the birth of the Solar System, raises the question whether a non-negligible amount of formamide may have been exogenously delivered onto a very young Earth about four billion years ago. A crucial part of the effort to 1 answer this question involves searching for formamide in regions where stars and planets are forming today in our Galaxy, as this can shed light on its formation, survival, and chemical re-processing along the different evolutionary phases leading to a star and planetary system like our own. -

Systematic Approach for Chemical Reactivity
SYSTEMATIC APPROACH FOR CHEMICAL REACTIVITY EVALUATION A Dissertation by ABDULREHMAN AHMED ALDEEB Submitted to the Office of Graduate Studies of Texas A&M University in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY December 2003 Major Subject: Chemical Engineering SYSTEMATIC APPROACH FOR CHEMICAL REACTIVITY EVALUATION A Dissertation by ABDULREHMAN AHMED ALDEEB Submitted to the Office of Graduate Studies of Texas A&M University in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY Approved as to style and content by: ____________________________ ____________________________ M. Sam Mannan Kenneth R. Hall (Chair of Committee) (Member) ____________________________ ____________________________ Mark T. Holtzapple Jerald A. Caton (Member) (Member) ____________________________ Kenneth R. Hall (Head of Department) December 2003 Major Subject: Chemical Engineering iii ABSTRACT Systematic Approach for Chemical Reactivity Evaluation. (December 2003) Abdulrehman Ahmed Aldeeb, B.S., Jordan University of Science & Technology; M.S., The University of Texas at Arlington Chair of Advisory Committee: Dr. M. Sam Mannan Under certain conditions, reactive chemicals may proceed into uncontrolled chemical reaction pathways with rapid and significant increases in temperature, pressure, and/or gas evolution. Reactive chemicals have been involved in many industrial incidents, and have harmed people, property, and the environment. Evaluation of reactive chemical hazards is critical to design and operate safer chemical plant processes. Much effort is needed for experimental techniques, mainly calorimetric analysis, to measure thermal reactivity of chemical systems. Studying all the various reaction pathways experimentally however is very expensive and time consuming. Therefore, it is essential to employ simplified screening tools and other methods to reduce the number of experiments and to identify the most energetic pathways. -

Introduction to NMR Spectroscopy of Proteins
A brief introduction to NMR spectroscopy of proteins By Flemming M. Poulsen 2002 1 Introduction Nuclear magnetic resonance, NMR, and X-ray crystallography are the only two methods that can be applied to the study of three-dimensional molecular structures of proteins at atomic resolution. NMR spectroscopy is the only method that allows the determination of three-dimensional structures of proteins molecules in the solution phase. In addition NMR spectroscopy is a very useful method for the study of kinetic reactions and properties of proteins at the atomic level. In contrast to most other methods NMR spectroscopy studies chemical properties by studying individual nuclei. This is the power of the methods but sometimes also the weakness. NMR spectroscopy can be applied to structure determination by routine NMR techniques for proteins in the size range between 5 and 25 kDa. For many proteins in this size range structure determination is relatively easy, however there are many examples of structure determinations of proteins, which have failed due to problems of aggregation and dynamics and reduced solubility. It is the purpose of these notes to introduce the reader to descriptions and applications of the methods of NMR spectroscopy most commonly applied in scientific studies of biological macromolecules, in particular proteins. The figures 1,2 and 11 are copied from “Multidímensional NMR in Liquids” by F.J.M de Ven (1995)Wiley-VCH The Figure13 and Table 1 have been copied from “NMR of Proteins and Nucleic acis” K. Wüthrich (1986) Wiley Interscience The Figures 20, 21 and 22 have been copied from J. -

Chapter 6 – the Structure and Reactivity of Molecules the Structure of Molecules This Is the Golden Age of Instrumentation
Chapter 6 – The Structure and Reactivity of Molecules The Structure of Molecules This is the golden age of instrumentation. Many of the techniques you used in organic lab were used a hundred years ago as the sole method of structure determination. Remember the nucleus has been known for just over 100 years! Beginning in the 1920's and 30's through today, we have seen the invention and the development of many analytical techniques used in structure determination. Infrared, NMR, UV-visible (electronic), and ESR spectroscopies, electrochemistry, X-ray crystallography are the major ones. Sixty years ago, an X-ray crystal structure could be an entire Ph.D. dissertation. Today an undergraduate can frequently solve the same structure in a single afternoon. We will talk about some of these methods later in the chapter. Valence Shell Electron Pair Repulsion (VSEPR) Theory While determining molecular structure is increasingly simple, a simple method of predicting structure is still invaluable. The method of choice is VSEPR theory. You saw this in CHM 211 so we'll only briefly review it. This method is a little different from the book’s and can be found in my general chemistry notes. Movable images of the different shapes can be found at http://science.marshall.edu/castella/chm211/vsepr.html (You will need Java® to view this page. Also you will have to use the Java® control panel to add this site to the secure list.). 1) Draw the Lewis structure of the molecule. 2) Determine the base shape of the molecule by adding the number of atoms bound to the central atom to the number of lone pairs on the central atom. -

Chapter 1: Atoms, Molecules and Ions
Previous Chapter Table of Contents Next Chapter Chapter 1: Atoms, Molecules and Ions Section 1.1: Introduction In this course, we will be studying matter, “the stuff things are made of”. There are many ways to classify matter. For instance, matter can be classified according to the phase, that is, the physical state a material is in. Depending on the pressure and the temperature, matter can exist in one of three phases (solid, liquid, or gas). The chemical structure of a material determines the range of temperatures and pressures under which this material is a solid, a liquid or a gas. Consider water for example. The principal differences between water in the solid, liquid and gas states are simply: 1) the average distance between the water molecules; small in the solid and the liquid and large in the gas and 2) whether the molecules are organized in an orderly three-dimensional array (solid) or not (liquid and gas). Another way to classify matter is to consider whether a substance is pure or not. So, matter can be classified as being either a pure substance or a mixture. A pure substance has unique composition and properties. For example, water is a pure substance (whether from Texas or Idaho, each water molecule always contains 2 atoms of hydrogen for 1 atom of oxygen). Under the same atmospheric pressure and at the same ambient temperature, water always has the same density. We can go a little further and classify mixtures are either homogeneous or heterogeneous. In a homogeneous mixture, for example, as a result of mixing a teaspoon of salt in a glass of water, the composition of the various components and their properties are the same throughout. -

Interpreting the Periodic Table – Teacher Page
Name: _____________________ Period: ____ Interpreting the Periodic Table – Teacher Page TEKS 8.5 Matter and energy. The student knows that matter is composed of atoms and has chemical and physical properties. The student is expected to: (C) interpret the arrangement of the Periodic Table, including groups and periods, to explain how properties are used to classify elements; Pre-Teach This is an activity designed to help students demonstrate an understanding of the arrangement of the periodic table. Students should be familiar with how the periodic table is arranged by atomic number, mass, energy levels, groups, periods, reactivity, valence electrons, reactivity etc. through other forms of presentation before completing this activity. A good activity to help construct this information for students would be the Mr. X activity. Differentiation Having students work together on this activity can be beneficial in some instances, but can also hinder student’s recall of the information. This is intended to be a summative assessment for this particular TEK before moving on to another concept. It is recommended that the assignment be modified so that the patterns could be easier to identify by reducing the number of letters on the “hypothetical periodic table” and strategically placing them on the table so they are closer together. Name: _____________________ Period: ____ Interpreting the Periodic Table Examine the hypothetical periodic table shown below. Use this periodic table to answer the questions that follow. B A D E C G F I H 1. Which pair(s) of elements has the same number of valence electrons? _____________________________________________________________ 2. How many valence electrons do they have? _____________________________________________________________ 3. -

Electron Paramagnetic Resonance of Radicals and Metal Complexes. 2. International Conference of the Polish EPR Association. Wars
! U t S - PL — voZ, PL9700944 Warsaw, 9-13 September 1996 ELECTRON PARAMAGNETIC RESONANCE OF RADICALS AND METAL COMPLEXES 2nd International Conference of the Polish EPR Association INSTITUTE OF NUCLEAR CHEMISTRY AND TECHNOLOGY UNIVERSITY OF WARSAW VGL 2 8 Hi 1 2 ORGANIZING COMMITTEE Institute of Nuclear Chemistry and Technology Prof. Andrzej G. Chmielewski, Ph.D., D.Sc. Assoc. Prof. Hanna B. Ambroz, Ph.D., D.Sc. Assoc. Prof. Jacek Michalik, Ph.D., D.Sc. Dr Zbigniew Zimek University of Warsaw Prof. Zbigniew Kqcki, Ph.D., D.Sc. ADDRESS OF ORGANIZING COMMITTEE Institute of Nuclear Chemistry and Technology, Dorodna 16,03-195 Warsaw, Poland phone: (0-4822) 11 23 47; telex: 813027 ichtj pi; fax: (0-4822) 11 15 32; e-mail: [email protected] .waw.pl Abstracts are published in the form as received from the Authors SPONSORS The organizers would like to thank the following sponsors for their financial support: » State Committee of Scientific Research » Stiftung fur Deutsch-Polnische Zusammenarbeit » National Atomic Energy Agency, Warsaw, Poland » Committee of Chemistry, Polish Academy of Sciences, Warsaw, Poland » Committee of Physics, Polish Academy of Sciences, Poznan, Poland » The British Council, Warsaw, Poland » CIECH S.A. » ELEKTRIM S.A. » Broker Analytische Messtechnik, Div. ESR/MINISPEC, Germany 3 CONTENTS CONFERENCE PROGRAM 9 LECTURES 15 RADICALS IN DNA AS SEEN BY ESR SPECTROSCOPY M.C.R. Symons 17 ELECTRON AND HOLE TRANSFER WITHIN DNA AND ITS HYDRATION LAYER M.D. Sevilla, D. Becker, Y. Razskazovskii 18 MODELS FOR PHOTOSYNTHETIC REACTION CENTER: STEADY STATE AND TIME RESOLVED EPR SPECTROSCOPY H. Kurreck, G. Eiger, M. Fuhs, A Wiehe, J. -

Trends in Reactivity in the Periodic Table
Chemistry for the gifted and talented 35 Trends in reactivity in the Periodic Table Student worksheet: CDROM index 18SW Discussion of answers: CDROM index 18DA Topics Trends in reactivity of Groups 1 and 7 and the ionic bonding model. Level Very able students aged 14–16. Prior knowledge Ionic bonding and the Periodic Table. Rationale This activity aims to: • help students develop a tool (flowcharts) to aid organisation of their line of reasoning; • help students explore links between trends in the reactivity of Groups 1 and 7 and atomic structure; • give students the opportunity to use and critically evaluate the relevance of ionisation energy data to the reactivity series of metals; •reinforce the idea of chemical changes being driven by energy changes; and • challenge the idea that Group 1 metals want to give away their outer shell electron. Use This could be used to follow up some work on the Periodic Table where the trends in reactivity in Groups 1 and 7 have been identified. It can be used as a differentiated activity for the more able students within a group. When the students have completed the worksheet they should be given the Discussion of answers sheet. They could check their own work or conduct a peer review of the work of Chemistry for the gifted and talented Trends in reactivity in the Periodic Table Flow charts are sometimes a good way to organise your thoughts into a line of reasoning or logical argument. The example below is about the question of why Group 1 elements get more reactive as you go down the group. -

How to Read and Interpret FTIR Spectroscope of Organic Material
97| Indonesian JournalIndonesian of Science Journal & Technology of Science &,Volum Technologye 4 Issue 4 ( 11,) (20April19 )2019 97-1 18Page 97-118 Indonesian Journal of Science & Technology Journal homepage: http://ejournal.upi.edu/index.php/ijost/ How to Read and Interpret FTIR Spectroscope of Organic Material Asep Bayu Dani Nandiyanto, Rosi Oktiani, Risti Ragadhita Departemen Kimia, Universitas Pendidikan Indonesia, Jl. Dr. Setiabudi no 229 Bandung 40154, Indonesia Correspondence: E-mail: [email protected] A B S T R A C T S A R T I C L E I N F O Article History: Fourier Transform Infrared (FTIR) has been developed as a Received 10 Jan 2019 tool for the simultaneous determination of organic Revised 20 Jan 2019 components, including chemical bond, as well as organic Accepted 31 Aug 2019 Available online 09 Apr 2019 content (e.g., protein, carbohydrate, and lipid). However, ____________________ until now, there is no further documents for describing the Keywords: detailed information in the FTIR peaks. The objective of this FTIR, study was to demonstrate how to read and assess chemical infrared spectrum, organic material, bond and structure of organic material in the FTIR, in which chemical bond, the analysis results were then compared with the literatures. organic structure. The step-by-step method on how to read the FTIR data was also presented, including reviewing simple to the complex organic materials. This study is potential to be used as a standard information on how to read FTIR peaks in the biochemical and organic materials. © 2019Tim Pengembang Jurnal UPI 1. INTRODUCTION analysis is quite rapid, good in accuracy, and relatively sensitive (Jaggi and Vij, 2006). -

Numerical Methods in Electron Magnetic Resonance
NO0000039 NUMERICAL METHODS IN ELECTRON MAGNETIC RESONANCE ANALYSES AND SIMULATIONS IN QUEST OF STRUCTURAL AND DYNAMICAL PROPERTIES OF FREE RADICALS IN SOLIDS BY ANDERS RASMUS S0RNES Thesis submitted for the degree Doctor scientiarum Department of Physics, University of Oslo 1998 31-18 Preface This thesis encompasses a collection of six theoretical and experimental Electron Magnetic Resonance (EMR) studies of the structure and dynamics of free radicals in solids. Radicals are molecular systems having one or more unpaired electron. Due to their energetically unfavourable open shell configuration, they are highly reactive. Radicals are formed in different ways. They are believed to be present in many chemical reactions as short-lived intermediate products, and they are also produced in interactions of x-radiation with matter, in Compton and photoelectric processes and interactions of their secondary electrons. Longer-lived, quasistable radicals do also exist and these may be used as spin-labels or spin-probes and may be attached to larger molecules and surfaces as measurement probes to examine the environment in their immediate vicinity. EMR is a collective term for a group of spectroscopic techniques inducing and observing transitions between the Zeeman levels of a paramagnetic system, such as a radical situated in an external magnetic field. Whithin the broader concept of EMR, the particular experimental techniques of Electron Paramagnetic Resonance (EPR), ELectron Nuclear DOuble Resonance (ENDOR), Field Swept ENDOR (FSE) and Electron Spin Echo Envelope Modulation (ESEEM) are discussed in this thesis. The focal point of this thesis is the development and use of numerical methods in the analysis, simulation and interpretation of EMR experiments on radicals in solids to uncover the structure, the dynamics and the environment of the system. -
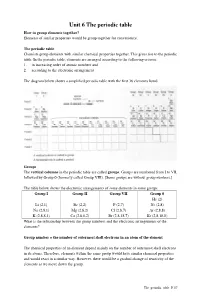
Unit 6 the Periodic Table How to Group Elements Together? Elements of Similar Properties Would Be Group Together for Convenience
Unit 6 The periodic table How to group elements together? Elements of similar properties would be group together for convenience. The periodic table Chemists group elements with similar chemical properties together. This gives rise to the periodic table. In the periodic table, elements are arranged according to the following criteria: 1. in increasing order of atomic numbers and 2. according to the electronic arrangement The diagram below shows a simplified periodic table with the first 36 elements listed. Groups The vertical columns in the periodic table are called groups . Groups are numbered from I to VII, followed by Group 0 (formerly called Group VIII). [Some groups are without group numbers.] The table below shows the electronic arrangements of some elements in some groups. Group I Group II Group VII Group 0 He (2) Li (2,1) Be (2,2) F (2,7) Ne (2,8) Na (2,8,1) Mg (2,8,2) Cl (2,8,7) Ar (2,8,8) K (2,8,8,1) Ca (2,8,8,2) Br (2,8,18,7) Kr (2,8,18,8) What is the relationship between the group numbers and the electronic arrangements of the elements? Group number = the number of outermost shell electrons in an atom of the element The chemical properties of an element depend mainly on the number of outermost shell electrons in its atoms. Therefore, elements within the same group would have similar chemical properties and would react in a similar way. However, there would be a gradual change of reactivity of the elements as we move down the group.