New Sesquiterpene Lactones from the Genera Calea and Berlandiera (Asteraceae) and the Fragmentation Reactions of 1,3-Dihydroxyeudesmanolide Derivatives
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Download Download
IN MEMORIAM: DONALD J. PINKAVA (29 AUGUST 1933–25 JULY 2017) Liz Makings Herbarium (ASU), School of Life Sciences Arizona State University, P.O. Box 874108 Tempe, Arizona 85287-4108, U.S.A. [email protected] My name is Liz Makings and I am the collections manager of the Arizona State University Herbarium. I was a graduate student at ASU in 2000 when I met Dr. Pinkava and he had just retired, so while I missed out on his talents as a teacher, I was lucky to get to know him as a mentor, colleague, and friend. Dr. Pinkava had a heart of gold, a mind like a trap, and a delightful collection of idiosyncrasies that was perfectly suited to his career path. He was hired at ASU in 1964 after completing his PhD. at Ohio State and was immediately responsible for teaching a 300 level botany class called “Flora of Arizona.” He undertook this responsibility with a meticulousness and attention to detail that can only be described as “Pinkavesque,” col- lecting the plants, learning the flora, and scouring the state for the best field trip sites. To his students he was simultaneously feared and adored. His exams turned men into boys and triggered anxiety attacks even among the best. He did not give grades, you earned them. There was no one more demanding, no one more thorough, yet no one more caring and helpful. Many former students have sung his praises and I’ll share this quote from one: “Dr. Pinkava was one of the kindest scientists I have ever interacted with, a trait that sometimes goes missing in our academic world. -

Chromosome Numbers in Compositae, XII: Heliantheae
SMITHSONIAN CONTRIBUTIONS TO BOTANY 0 NCTMBER 52 Chromosome Numbers in Compositae, XII: Heliantheae Harold Robinson, A. Michael Powell, Robert M. King, andJames F. Weedin SMITHSONIAN INSTITUTION PRESS City of Washington 1981 ABSTRACT Robinson, Harold, A. Michael Powell, Robert M. King, and James F. Weedin. Chromosome Numbers in Compositae, XII: Heliantheae. Smithsonian Contri- butions to Botany, number 52, 28 pages, 3 tables, 1981.-Chromosome reports are provided for 145 populations, including first reports for 33 species and three genera, Garcilassa, Riencourtia, and Helianthopsis. Chromosome numbers are arranged according to Robinson’s recently broadened concept of the Heliantheae, with citations for 212 of the ca. 265 genera and 32 of the 35 subtribes. Diverse elements, including the Ambrosieae, typical Heliantheae, most Helenieae, the Tegeteae, and genera such as Arnica from the Senecioneae, are seen to share a specialized cytological history involving polyploid ancestry. The authors disagree with one another regarding the point at which such polyploidy occurred and on whether subtribes lacking higher numbers, such as the Galinsoginae, share the polyploid ancestry. Numerous examples of aneuploid decrease, secondary polyploidy, and some secondary aneuploid decreases are cited. The Marshalliinae are considered remote from other subtribes and close to the Inuleae. Evidence from related tribes favors an ultimate base of X = 10 for the Heliantheae and at least the subfamily As teroideae. OFFICIALPUBLICATION DATE is handstamped in a limited number of initial copies and is recorded in the Institution’s annual report, Smithsonian Year. SERIESCOVER DESIGN: Leaf clearing from the katsura tree Cercidiphyllumjaponicum Siebold and Zuccarini. Library of Congress Cataloging in Publication Data Main entry under title: Chromosome numbers in Compositae, XII. -

Illinois Bundleflower (Desmanthus Illinoensis) Story by Alan Shadow, Manager USDA-NRCS East Texas Plant Materials Center Nacogdoches, Texas
Helping People Help The Land September/October 2011 Issue No. 11 The Reverchon Naturalist Recognizing the work of French botanist Julien Reverchon, who began collecting throughout the North Central Texas area in 1876, and all the botanists/naturalists who have followed ... Drought, Heat and Native Trees ranging from simple things like more extensive root systems, to more drastic measures like pre- Story by Bruce Kreitler mature defoliation, what they actually have little Abilene, Texas defense against is a very prolonged period of no appreciable water supply. nybody that has traveled in Texas this year A will have noticed that not only most of the By the way, even though they are usually the land browned out, but also if you look at the trees same species, there is a difference in landscape in the fields and beside the roads, they aren't trees and native trees, which are untended plants looking so good either. It doesn't take a rocket that have to fend for themselves. While they are scientist to realize that extreme high temperatures indeed the same basic trees, the differences be- combined with, and partially caused by, drought tween the environments that they live in are huge are hard on trees. and thus overall general environmental factors such as drought, temperature, and insect infesta- Since I'm pretty sure that most of the people read- tions act on them differently. For the purposes of ing this article understand very well that drought this article, I'm referring to trees that are on their is a problem for trees, the question isn't is the pre- own, untended for their entire lives in fields, pas- sent drought going to have an effect on trees, but tures, forests, or just wherever nature has placed rather, what are the present effects of the drought them and refer to them as native trees. -

Earth-Kind® Perennial Plant List
Earth-Kind® Perennial Plant Research Garden at Myers Park: McKinney, Texas Scientific Name Common Name 1 Achillea millefolium ' Moonshine' Yarrow 2 Agastache aurantiaca ' Apricot Sunrise' Hyssop or Hummingbird Mint 3 Anisacanthus quadrifidus var. wrightii Flame Acanthus 4 Aster oblongifolia Aromatic Aster 5 Baptisia australis ' Big Burly' False Indigo 6 Berlandiera lyrata Chocolate Daisy 7 Calylophus drummondianus 'Berlandiera' Texas Primrose - Compact Gold 8 Chrysactinia mexicana Damianita Daisy 9 Chrysanthemum leucanthemum Ox-eye Daisy 10 Eupatorium greggii Gregg's Mistflower Texas AgriLife Extension Service and Collin County Master Gardeners 1 Earth-Kind® Perennial Plant Research Garden at Myers Park: McKinney, Texas Scientific Name Common Name 11 Datura wrightii Angel Trumpet 12 Delosperma cooperi Ice Plant 13 Dianthus gratianopolitanus ' Firewitch' Firewitch Cheddar Pink 14 Dianthus ' First Love' First Love Dianthus 15 Echinacea purpurea ' Kim's Knee High' Purple Coneflower - Kim's Knee High 16 Echinacea purpurea ' White Swan' White Coneflower 17 Engelmannia pinnatifida Englemann Daisy, Cutleaf Daisy 18 Eupatorium purpureum Joe Pye Weed 19 Gaura lindheimeri ' Siskiyou Pink' Whirling Butterflies 'Siskiyou Pink' 20 Gaura lindheimeri 'Pink Cloud' Whirling Butterflies 'Pink Cloud' Texas AgriLife Extension Service and Collin County Master Gardeners 2 Earth-Kind® Perennial Plant Research Garden at Myers Park: McKinney, Texas Scientific Name Common Name 21 Gaura lindheimeri ' White Fountain' Whirling Butterflies 'White Fountain' 22 Hemerocallis -

Vascular Plants and a Brief History of the Kiowa and Rita Blanca National Grasslands
United States Department of Agriculture Vascular Plants and a Brief Forest Service Rocky Mountain History of the Kiowa and Rita Research Station General Technical Report Blanca National Grasslands RMRS-GTR-233 December 2009 Donald L. Hazlett, Michael H. Schiebout, and Paulette L. Ford Hazlett, Donald L.; Schiebout, Michael H.; and Ford, Paulette L. 2009. Vascular plants and a brief history of the Kiowa and Rita Blanca National Grasslands. Gen. Tech. Rep. RMRS- GTR-233. Fort Collins, CO: U.S. Department of Agriculture, Forest Service, Rocky Mountain Research Station. 44 p. Abstract Administered by the USDA Forest Service, the Kiowa and Rita Blanca National Grasslands occupy 230,000 acres of public land extending from northeastern New Mexico into the panhandles of Oklahoma and Texas. A mosaic of topographic features including canyons, plateaus, rolling grasslands and outcrops supports a diverse flora. Eight hundred twenty six (826) species of vascular plant species representing 81 plant families are known to occur on or near these public lands. This report includes a history of the area; ethnobotanical information; an introductory overview of the area including its climate, geology, vegetation, habitats, fauna, and ecological history; and a plant survey and information about the rare, poisonous, and exotic species from the area. A vascular plant checklist of 816 vascular plant taxa in the appendix includes scientific and common names, habitat types, and general distribution data for each species. This list is based on extensive plant collections and available herbarium collections. Authors Donald L. Hazlett is an ethnobotanist, Director of New World Plants and People consulting, and a research associate at the Denver Botanic Gardens, Denver, CO. -

Relative Ranking of Ornamental Flower Plants to Foraging Honey Bees (With Notes on Favorability to Bumble Bees)
Relative Ranking of Ornamental Flower Plants to Foraging Honey Bees (With Notes on Favorability to Bumble Bees) Whitney Cranshaw Colorado State University Observations were made during the 2007-2009 growing seasons on the relative attractiveness of various flowering ornamental plants to honey bees (Apis mellifera). This information was collected so that honey bee favorability - or lack of favorability - may be considered in plant selection. The study was conducted by repeated visits to public garden plantings in Larimer, Denver, Adams, and Cheyenne counties. Gardens were chosen that had large mass plantings of numerous flowering plants so that comparisons could be made and included the Denver Botanic Garden, gardens at Colorado State University (PERC, Flower Demonstration Planting), Welby Gardens, and Cheyenne Botanic Garden. These sites also were chosen because plantings had identification labeling. Plantings were visited between 2 and 12 times between mid-June and mid-September. Evaluations were made by examining plants that were in flower for the presence of honey bees. A planting was then given a relative ranking based on honey bee numbers. A 0-3 scale was used: 3 - Heavily visited by foraging honey bees 2 - Moderately visited by honey bees and foraged 1 - Honey bees seen occasionally visiting flowers 0 - Honey bees do not forage at these flowers Data were collected from a total of 319 different plant entries durig this study. Variation in rankings between dates did occur; where this occurred from multiple ratings the final ranking was rounded up to a whole number. Numerous other bees and other insects were commonly seen on many plants. -

University Microfilms, Inc., Ann Arbor, Michigan BIOSYSTEMATIC STUDY of OENUS BERLANDIERA DC
This dissertation has been 65—3903 microfilmed exactly as received PINKAVA, Donald John, 1933- BXDSYSTEMATIC STUDY OF GENUS BERLANDIERA DC. (COMPOS1TAE). The Ohio State University, Ph.D., 1964 Botany University Microfilms, Inc., Ann Arbor, Michigan BIOSYSTEMATIC STUDY OF OENUS BERLANDIERA DC. (COMPOSITAE) DISSERTATION Presented in Partial Fulfillment of tha Requirement* for the Degree Doctor of Philosophy in the Graduate School of The Ohio State University By DONALD JOHN PINKAVA, B.Sc.t M.Sc. The Ohio State University 1964 Approved by Adviser Department of Botany and Plant Pathology PLEASE NOTE Figure pages are not original copy. They tend to "curl". Filmed in the best possible way. University Microfilms, Inc. ACKNOWLEDGMENTS I wish to express my sincere gratitude to Dr* T. Richard Fisher, my adviser, who not only suggested thia problem, but moat effectively guided it to ita completion* I am thankful alao to Drs* Clara G. Weiahaupt, Emanuel 0. Rudolph, Dale A. Ray and Carroll A* Swanson for reading thia dissertation and for their helpful criticisms and suggestions* Special recognition is extended to John M* Speer* Unless otherwise cited, the photography is to be credited to his talents so unselfishly shared* I am deeply indebted to Dr* Ray for statistical assistance; to the curators of the herbaria for loaned specimens and/or photographs; and to my colleagues for their many useful suggestions* Financial assistance was provided by The Ohio State Research Foundation and the National Science Foundation* ii TABLE OF CONTENTS Pag* ACKNOWLEDGMENTS....................................... 11 LIST OF T A B I E S ......................................... It LIST OF ILLUSTRATIONS................................. Yl INTRODUCTION........................................... 1 GENUS D E S C R I P T I O N .................................... -

Appendix A. Plant Species Known to Occur at Canaveral National Seashore
National Park Service U.S. Department of the Interior Natural Resource Stewardship and Science Vegetation Community Monitoring at Canaveral National Seashore, 2009 Natural Resource Data Series NPS/SECN/NRDS—2012/256 ON THE COVER Pitted stripeseed (Piriqueta cistoides ssp. caroliniana) Photograph by Sarah L. Corbett. Vegetation Community Monitoring at Canaveral National Seashore, 2009 Natural Resource Report NPS/SECN/NRDS—2012/256 Michael W. Byrne and Sarah L. Corbett USDI National Park Service Southeast Coast Inventory and Monitoring Network Cumberland Island National Seashore 101 Wheeler Street Saint Marys, Georgia, 31558 and Joseph C. DeVivo USDI National Park Service Southeast Coast Inventory and Monitoring Network University of Georgia 160 Phoenix Road, Phillips Lab Athens, Georgia, 30605 March 2012 U.S. Department of the Interior National Park Service Natural Resource Stewardship and Science Fort Collins, Colorado The National Park Service, Natural Resource Stewardship and Science office in Fort Collins, Colorado publishes a range of reports that address natural resource topics of interest and applicability to a broad audience in the National Park Service and others in natural resource management, including scientists, conservation and environmental constituencies, and the public. The Natural Resource Data Series is intended for the timely release of basic data sets and data summaries. Care has been taken to assure accuracy of raw data values, but a thorough analysis and interpretation of the data has not been completed. Consequently, the initial analyses of data in this report are provisional and subject to change. All manuscripts in the series receive the appropriate level of peer review to ensure that the information is scientifically credible, technically accurate, appropriately written for the intended audience, and designed and published in a professional manner. -

Project Budburst Available Species Sheet
Project BudBurst Available Species Sheet www.budburst.org Wildflowers and Herbs Deciduous Trees and Shrubs • Alfalfa (Medicago sativa) • American linden (Tilia americana) • American pasqueflower (Pulsatilla patens aka • Antelope bitterbrush (Purshia tridentata) Anemone patens) • Apple (Malus pumila) • Bigleaf lupine (Lupinus polyphyllus) • Bald cypress (Taxodium distichum) • Bitter root (Lewisia rediviva) • Balsam poplar (Populus balsamifera (aka • California poppy (Eschscholzia californica) trichocarpa)) • Canada thistle (Cirsium arvense) • Beaked hazelnut (Corylus cornuta) • Colorado blue columbine (Aquilegia caerulea) • Bigleaf maple (Acer macrophyllum) • Common dandelion (Taraxacum officinale) • Black elderberry (Sambucus nigra) • Common yarrow (Achillea millefolium) • Black locust (Robinia pseudoacacia) • Darkthroat shootingstar (Dodecatheon • Boxelder (Acer negundo) pulchellum) • Chokecherry (Prunus virginiana) • Dogtooth violet (Erythronium americanum) • Common lilac (Syringa vulgaris) • Field mustard (Brassica rapa) • Common snowberry (Symphoricarpos albus) • Henbit deadnettle (Lamium amplexicaule) • Eastern serviceberry (Amelanchier canadensis) • Indian pink (Spigelia marilandica) • Flowering dogwood (Cornus florida) • Jack in the pulpit (Arisaema triphyllum) • Forsythia (Forsythia xintermedia) • Lanceleaf springbeauty (Claytonia lanceolata) • Lewis' mock orange (Philadelphus lewisii) • Large flowered trillium (Trillium grandiflorum) • Pacific dogwood (Cornus nuttallii) • Mayapple (Podophyllum peltatum) • Paper birch (Betula -

The Seed Plants, Ferns, and Fern Allies of the Austin Region
University of Texas Bulletin No. 2065: November 20, 1920 The Seed Plants, Ferns, and Fern Allies of the Austin Region By Mary Sophie Young, Ph.D. PUBLISHED BY THB UNIVERSITY OF TBXAS AUSTIN : Publications of the University of Texas Publications Committee Frederic Duncalf C. T. Gray Killis Campbell E. J. Mathews D. B. Casteel C. E. Rowe F. W. Graff A. E. Trombly The University publishes bulletins six times a month, so numbered that the first two digits of the number show the year of issue, the last two the position in the yearly series. (For example, No. 1701 is the first bulletin of the year 1917.) These comprise the official publications of the University, publications on humanistic and scientific sub- jects, bulletins prepared by the Bureau of Extension, by the Bureau of Government Research, and by the Bureau of Eco- nomic Geology and Technology, and other bulletins of gen- eral educational interest. With the exception of special num- bers, any bulletin will be sent to a citizen of Texas free on request. All communications about University publications should be addressed to University Publications, University of Texas, Austin. 12-29-20-1357-6274-lm University of Texas Bulletin No. 2065: November 20, 1920 The Seed Plants, Ferns, and Fern Allies of the Austin Region By Mary Sophie ^ oung, Ph.D. PUBLISHED BY THE UNIVERSITY SIX TIMES A MONTH, AND ENTERED AS] SECOND-CLASS MATTER AT THE POSTOFFICE AT AUSTIN. TEXAS. UNDBR THE ACT OF AUGUST 24, 1912 v i |1*o The benefits of education and of useful knowledge, generally diffused through a community, are essential to the preservation of a free govern- ment. -
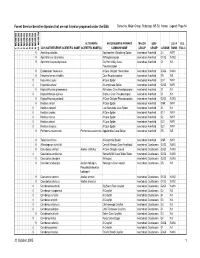
Sensitive Species That Are Not Listed Or Proposed Under the ESA Sorted By: Major Group, Subgroup, NS Sci
Forest Service Sensitive Species that are not listed or proposed under the ESA Sorted by: Major Group, Subgroup, NS Sci. Name; Legend: Page 94 REGION 10 REGION 1 REGION 2 REGION 3 REGION 4 REGION 5 REGION 6 REGION 8 REGION 9 ALTERNATE NATURESERVE PRIMARY MAJOR SUB- U.S. N U.S. 2005 NATURESERVE SCIENTIFIC NAME SCIENTIFIC NAME(S) COMMON NAME GROUP GROUP G RANK RANK ESA C 9 Anahita punctulata Southeastern Wandering Spider Invertebrate Arachnid G4 NNR 9 Apochthonius indianensis A Pseudoscorpion Invertebrate Arachnid G1G2 N1N2 9 Apochthonius paucispinosus Dry Fork Valley Cave Invertebrate Arachnid G1 N1 Pseudoscorpion 9 Erebomaster flavescens A Cave Obligate Harvestman Invertebrate Arachnid G3G4 N3N4 9 Hesperochernes mirabilis Cave Psuedoscorpion Invertebrate Arachnid G5 N5 8 Hypochilus coylei A Cave Spider Invertebrate Arachnid G3? NNR 8 Hypochilus sheari A Lampshade Spider Invertebrate Arachnid G2G3 NNR 9 Kleptochthonius griseomanus An Indiana Cave Pseudoscorpion Invertebrate Arachnid G1 N1 8 Kleptochthonius orpheus Orpheus Cave Pseudoscorpion Invertebrate Arachnid G1 N1 9 Kleptochthonius packardi A Cave Obligate Pseudoscorpion Invertebrate Arachnid G2G3 N2N3 9 Nesticus carteri A Cave Spider Invertebrate Arachnid GNR NNR 8 Nesticus cooperi Lost Nantahala Cave Spider Invertebrate Arachnid G1 N1 8 Nesticus crosbyi A Cave Spider Invertebrate Arachnid G1? NNR 8 Nesticus mimus A Cave Spider Invertebrate Arachnid G2 NNR 8 Nesticus sheari A Cave Spider Invertebrate Arachnid G2? NNR 8 Nesticus silvanus A Cave Spider Invertebrate Arachnid G2? NNR -

An Analysis of MOFEP Ground Flora
An Analysis of MOFEP Ground Flora: Pre-treatment Conditions Jennifer K. Grabner, David R. Larsen, and John M. Kabrick1 Abstract.-8imilarities and differences in MOFEP ground flora spe cies composition were determined at site, block, and treatment levels. Ground flora data were collected across nine sites on 648 permanent forestry plots; more than 10,300 1-m2 quadrats were sampled each summer from 1991 through 1995. Approximately 530 species were identified; more than half occurred on fewer than 10 percent of the plots. Highly significant differences among sample years were ob served for plot richness, but were regarded as a reflection of improved data quality over the course of the project. Though plots averaged relatively high species diversity, wide ranges from low to high species richness and diversity existed within all sites. Analysis of variance on plot diversity and richness indicated a strong trend of differences between even-aged and control sites. Differences in ground flora species composition and abundance, plot richness, and plot diversity appear strongly correlated with pattems in geology, landform, and soils both within and among the MOFEP sites. To practice effective ecosystem management, 1971 and 1976). To date, there has been no natural resource managers must develop an comprehensive evaluation of Missouri's upland understanding of relationships among the major Ozark ground flora. MOFEP provides an unpar components of the systems they are managing. alleled opportunity to thoroughly describe The Missouri Ozark Forest Ecosystem Project current ground flora conditions in mature (MOFEP) is a large-scale, long-term experiment second-growth oak/hickory and oak/pine to investigate effects of even-aged, uneven-aged, forests in the southeast Missouri Ozarks.