2549-2577.Pdf
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Use of Capillary Electrophoresis (CE) in Drug Analysis
Florida International University FIU Digital Commons FIU Electronic Theses and Dissertations University Graduate School 3-28-2005 The use of capillary electrophoresis (CE) in drug analysis Agnes D. Garcia Follow this and additional works at: https://digitalcommons.fiu.edu/etd Part of the Other Life Sciences Commons Recommended Citation Garcia, Agnes D., "The use of capillary electrophoresis (CE) in drug analysis" (2005). FIU Electronic Theses and Dissertations. 3928. https://digitalcommons.fiu.edu/etd/3928 This work is brought to you for free and open access by the University Graduate School at FIU Digital Commons. It has been accepted for inclusion in FIU Electronic Theses and Dissertations by an authorized administrator of FIU Digital Commons. For more information, please contact [email protected]. FLORIDA INTERNATIONAL UNIVERSITY Miami, Florida THE USE OF CAPILLARY ELECTROPHORESIS (CE) IN DRUG ANALYSIS A thesis submitted in partial fulfillment of the requirements for the degree of MASTER OF SCIENCE in FORENSIC SCIENCE by Agnes D. Garcia 2005 To: Dean R. Bruce Dunlap College of Arts and Sciences This thesis, written by Agnes D. Garcia, and entitled The Use of Capillary Electrophoresis (CE) in Drug Analysis, having been approved in respect to style and intellectual content, is referred to you for judgment. We have read this thesis and recommend that it be approved. Kenneth Furton Bruce McCord Jose R. Almirall, Major Professor Date of Defense: March 28,2005 The thesis of Agnes D. Garcia is approved. Dean R. Bruce Dunlap College of Arts and Sciences Dean Douglas Wartzok University Graduate School Florida International University, 2005 ii © Copyright 2005 by Agnes D. -

Testosterone Art. 31
Annex I List of the names, pharmaceutical forms, strengths of the medicinal products, routes of administration, and marketing authorisation holders in the Member States 1 Member Marketing authorisation INN Invented name Strength Pharmaceutical Route of State holder form administration (in EEA) Austria Bayer Austria GmbH testosterone undecanoat Nebido 1000 mg/4 ml 1000 mg/4ml solution for intramuscular use Herbststrasse 6-10 Injektionslösung injection 1160 Wien Austria Austria Eli Lilly GmbH testosterone Axiron 30 mg/1,5 ml 30 mg/1,5ml gel cutaneous use Kölblgasse 8-10 Lösung zur 1030 Wien Anwendung auf der Austria Haut Austria Laboratoires Besins testosterone Testogel 25 mg - Gel 25 mg gel cutaneous use International SA im Beutel Rue de Bourg lÀbbè 3 75003 Paris France Austria Laboratoires Besins testosterone Testogel 50 mg - Gel 50 mg gel cutaneous use International SA im Beutel Rue de Bourg lÀbbè 3 75003 Paris France Austria ProStrakan Ltd. testosterone Tostran 2% - Gel 0.02 gel cutaneous use Galabank Business Park TD1 1QH Galashiels United Kingdom Austria Laboratoires Besins testosterone Androgel 25 mg - Gel 25 mg gel cutaneous use International SA im Beutel Rue de Bourg lÀbbè 3 75003 Paris France Austria Laboratoires Besins testosterone Androgel 50 mg - Gel 50 mg gel cutaneous use International SA im Beutel Rue de Bourg lÀbbè 3 75003 Paris France Austria N.V. Organon testosterone undecanoat Andriol Testocaps 40 40 mg capsule, soft oral use Kloosterstraat 6 mg - Kapseln 5349 AB Oss The Netherlands 2 Member Marketing authorisation INN Invented name Strength Pharmaceutical Route of State holder form administration (in EEA) Belgium Besins Healthcare testosterone Androgel 25 mg gel transdermal use Avenue Louise 287 1050 Brussels Belgium Belgium Besins Healthcare testosterone Androgel 50 mg gel transdermal use Avenue Louise 287 1050 Brussels Belgium Belgium Eli Lilly Benelux S.A.-N.V. -

An Update the Increasing Popularity of Anabolic Androgenic Steroids
Anabolic Androgenic Steroid Abuse in the United Kingdom; An Update The increasing popularity of anabolic androgenic steroids. Carrie Mullena, Benjamin J Whalleyb, Fabrizio Schifanoc, Julien S Bakerd a School of Computing, Engineering and Physical Sciences, University of the West of Scotland, Paisley, PA1 2BE b School of Chemistry, Food and Nutritional Sciences, and Pharmacy, The University of Reading, Whiteknights, Reading, Berkshire, RG6 6AP c School of Life and Medical Sciences, University of Hertfordshire, Hatfield, Hertfordshire, AL10 9EU d Centre for Health and Exercise Science Research, Department of Sport, Physical Education and Health, Hong Kong Baptist University, Kowloon Tong, Hong Kong. Correspondence Carrie Mullen, School of Computing, Engineering and Physical Sciences, University of the West of Scotland, Paisley, PA1 2BE Tel 0044 141 848 3845 email [email protected] 7,081 Words Abstract Anabolic androgenic steroids (AASs) are prescribed for medical conditions related to low testosterone. Abuse of AASs has surged as they become increasingly recognised as potent image enhancement drugs. The primary goal of most abusers is to obtain what they consider to be a more attractive outward appearance. Abuse is complex. There are a vast range of AAS substances available, although due to their illicit nature, the true composition of AAS substances is difficult to evaluate. Users follow dosing patterns which incorporate a number This article has been accepted for publication and undergone full peer review but has not been through the copyediting, typesetting, pagination and proofreading process which may lead to differences between this version and the Version of Record. Please cite this article as doi: 10.1111/bph.14995 This article is protected by copyright. -

Use of High-Temperature Liquid Chromatography with Sub
This article was downloaded by: [Moskow State Univ Bibliote] On: 15 November 2013, At: 02:15 Publisher: Taylor & Francis Informa Ltd Registered in England and Wales Registered Number: 1072954 Registered office: Mortimer House, 37-41 Mortimer Street, London W1T 3JH, UK Journal of Liquid Chromatography & Related Technologies Publication details, including instructions for authors and subscription information: http://www.tandfonline.com/loi/ljlc20 Use of High-Temperature Liquid Chromatography with Sub-2 µm Particle C18 Columns for the Analysis of Seized Drugs Ira Lurie a & Li Li a a Special Testing and Research Laboratory, U.S. Drug Enforcement Administration , Dulles, Virginia, USA Published online: 05 Oct 2009. To cite this article: Ira Lurie & Li Li (2009) Use of High-Temperature Liquid Chromatography with Sub-2 µm Particle C18 Columns for the Analysis of Seized Drugs, Journal of Liquid Chromatography & Related Technologies, 32:18, 2615-2626, DOI: 10.1080/10826070903245516 To link to this article: http://dx.doi.org/10.1080/10826070903245516 PLEASE SCROLL DOWN FOR ARTICLE Taylor & Francis makes every effort to ensure the accuracy of all the information (the “Content”) contained in the publications on our platform. However, Taylor & Francis, our agents, and our licensors make no representations or warranties whatsoever as to the accuracy, completeness, or suitability for any purpose of the Content. Any opinions and views expressed in this publication are the opinions and views of the authors, and are not the views of or endorsed by Taylor & Francis. The accuracy of the Content should not be relied upon and should be independently verified with primary sources of information. -

(12) Patent Application Publication (10) Pub. No.: US 2008/0161324 A1 Johansen Et Al
US 2008O161324A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2008/0161324 A1 Johansen et al. (43) Pub. Date: Jul. 3, 2008 (54) COMPOSITIONS AND METHODS FOR Publication Classification TREATMENT OF VRAL DISEASES (51) Int. Cl. (76) Inventors: Lisa M. Johansen, Belmont, MA A63/495 (2006.01) (US); Christopher M. Owens, A63L/35 (2006.01) Cambridge, MA (US); Christina CI2O I/68 (2006.01) Mawhinney, Jamaica Plain, MA A63L/404 (2006.01) (US); Todd W. Chappell, Boston, A63L/35 (2006.01) MA (US); Alexander T. Brown, A63/4965 (2006.01) Watertown, MA (US); Michael G. A6II 3L/21 (2006.01) Frank, Boston, MA (US); Ralf A6IP3L/20 (2006.01) Altmeyer, Singapore (SG) (52) U.S. Cl. ........ 514/255.03: 514/647; 435/6: 514/415; Correspondence Address: 514/460, 514/275: 514/529 CLARK & ELBNG LLP 101 FEDERAL STREET BOSTON, MA 02110 (57) ABSTRACT (21) Appl. No.: 11/900,893 The present invention features compositions, methods, and kits useful in the treatment of viral diseases. In certain (22) Filed: Sep. 13, 2007 embodiments, the viral disease is caused by a single stranded RNA virus, a flaviviridae virus, or a hepatic virus. In particu Related U.S. Application Data lar embodiments, the viral disease is viral hepatitis (e.g., (60) Provisional application No. 60/844,463, filed on Sep. hepatitis A, hepatitis B, hepatitis C, hepatitis D, hepatitis E). 14, 2006, provisional application No. 60/874.061, Also featured are screening methods for identification of filed on Dec. 11, 2006. novel compounds that may be used to treat a viral disease. -

Appraisal of Test Methods for Sex-Hormone Disrupting Chemicals Capable of Affecting the Reproductive Process
APPRAISAL OF TEST METHODS FOR SEX-HORMONE DISRUPTING CHEMICALS CAPABLE OF AFFECTING THE REPRODUCTIVE PROCESS Prepared by the MRC Institute for Environment and Health for the UK Department of Environment, Transport and the Regions 1998 Prepared by the MRC Institute for Environment and Health Written by: Mr. P. Holmes Dr. C Humfrey Mr. M. Scullion With additional material from : Dr M Taylor 2 FOREWORD Over a number of years anecdotal observations and scientific evidence have come to light which suggest that chemicals present in the environment may cause adverse health effects in humans and wildlife because of their capacity to mimic or influence the action of endogenous hormones such as oestrogens and androgens. Current concerns centre around the apparent increase in human and animal disease states linked to the action of sex-hormones and reported observations from human epidemiological studies of reductions in sperm counts and fertility, increases in the incidence of testicular cancer and congenital birth defects in men and increases in the incidence of breast cancer in women. In wildlife species the occurrence of gross birth deformities, behavioral abnormalities and both feminisation and masculinisation are major concerns. While there remains much scientific uncertainty about the nature and magnitude of the human and environmental risks associated with exposure to endocrine disrupting chemicals, the concerns are such that in November 1996, OECD Member countries decided to work together on international projects to: • coordinate national and regional activities on the risk assessment and management of endocrine disrupting chemicals; • develop internationally acceptable methods for the hazard characterization of endocrine disrupting chemicals; and • harmonise risk characterization approaches and regulatory approaches. -

Identification of Unknown Residues
Project number: 871.639.01 BAS-code: WOT-02-438-III-036 Project title: Identification of unknown residues Project leader: R.J.B. Peters Report 2009.013 November 2009 Identification of Unknown Residues using bioassay directed fractionation, UPLC/TOFMS analysis and database searching R.J.B. Peters, J.C.W. Rijk, J.E. Oosterink, A.W.J.M. Nijrolder and M.W.F. Nielen Business Unit: Veterinary Drugs Group: Analytical Services & Development RIKILT - Institute of Food Safety Wageningen University and Research Centre Akkermaalsbos 2, 6708 WB Wageningen, The Netherlands P.O. Box 230, 6700 AE Wageningen, The Netherlands Tel +31 317 480 256 Fax +31 317 417 717 Internet www.rikilt.wur.nl Copyright 2009, RIKILT – Institute of Food Safety. The client is allowed to publish or distribute the full report to third parties. Without prior written permission from RIKILT – Institute of Food Safety it is not allowed to: a) publish parts of this report; b) use this report or title of this report in conducting legal procedures, for advertising, acquisition or other commercial purposes; c) use the name of RIKILT – Institute of Food Safety other than as author of this report. The research described in this report was funded by the Dutch Ministry of Agriculture, Nature and Food Quality (WOT-02-438-III-036) Distribution list: • Food and Consumer Product Safety Authority (VWA); J.A. van Rhijn • National Institute for Public Health and the Environment (RIVM); drs. M. Blokland, dr. L. van Ginkel, drs. S.S. Sterk This report from RIKILT - Institute of Food Safety has been produced with the utmost care. -

In Vitro Androgen Bioassays As a Detection Method for Designer Androgens
Sensors 2013, 13, 2148-2163; doi:10.3390/s130202148 OPEN ACCESS sensors ISSN 1424-8220 www.mdpi.com/journal/sensors Review In Vitro Androgen Bioassays as a Detection Method for Designer Androgens Elliot R. Cooper 1, Kristine C. Y. McGrath 1 and Alison K. Heather 1,2,* 1 School of Medical and Molecular Biosciences, University of Technology, Sydney, Ultimo 2007, New South Wales, Australia; E-Mails: [email protected] (E.R.C.); [email protected] (K.C.Y.M.) 2 Heart Research Institute, Newtown 2042, Sydney, New South Wales, Australia * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel.: +61-2-9514-4034; Fax: +61-2-9514-8206. Received: 6 December 2012; in revised form: 22 January 2013 / Accepted: 30 January 2013 / Published: 6 February 2013 Abstract: Androgens are the class of sex steroids responsible for male sexual characteristics, including increased muscle mass and decreased fat mass. Illicit use of androgen doping can be an attractive option for those looking to enhance sporting performance and/or physical appearance. The use of in vitro bioassays to detect androgens, especially designer or proandrogens, is becoming increasingly important in combating androgen doping associated with nutritional supplements. The nutritional sports supplement market has grown rapidly throughout the past decade. Many of these supplements contain androgens, designer androgens or proandrogens. Many designer or proandrogens cannot be detected by the standard highly-sensitive screening methods such as gas chromatography-mass spectrometry because their chemical structure is unknown. However, in vitro androgen bioassays can detect designer and proandrogens as these assays are not reliant on knowing the chemical structure but instead are based on androgen receptor activation. -
LGC Product List
Uni-Asia Scientific Instrument Company Limited - Your Worldwide Laboratory Supplies Sourcing Partner Email [email protected] Website www.uniasiahk.com Please browse through our product lists or send your enquiry to [email protected] for a quotation! Bulk pricing can always be discussed! LGC Standards Product List Code Discriptions AC-SE015.040.001 Drugs of substitution STM 3/10-A SE serum, 6 x 2.5ml AC-SE015.040.002 Drugs of substitution STM 1/11-A SE serum, 6 x 2.5ml AC-SE015.040.005.003TDM Benzodiazepine in serum, lyophilised BZF 3/10-A SE for AC-SE015.040.005.004TDM Benzodiazepine in serum, lyophilised BZF 3/10-B SE for AC-SE015.040.005.005TDM Benzodiazepine in serum, lyophilised BZF 1/11– A SE for AC-SE015.040.005.006TDM Benzodiazepine in serum, lyophilised BZF 1/11– B SE for BAM-K008 Diesel oil (ISO 9377-2, ISO 16703, EN 14039), 7ml BAM-K009 Lubricating oil (ISO 9377-2, ISO 16703, EN 14039), 7ml BAM-K010 Diesel Fuel/Lubricating Oil (1:1) (ISO 9377-2, ISO 16703, BAMSUS-1R Setting-up sample for sectrometric analysis of low alloyed BCR-010 Tin ore powder - Tin; 225g BCR-017A Copper - Sulfur and phosphorous, 370g BCR-017B Copper - Sulfur and phosphorous; 50g BCR-022A Copper (electrolytic tough pitch) - Oxygen; 42g BCR-022B Copper (electrolytic tough pitch) - Oxygen; 28g BCR-024B Titanium - Oxygen and nitrogen; 10g BCR-024C Titanium - Oxygen, nitrogen; 5g BCR-032 Moroccan phosphate rock - Trace elements; 100g BCR-033 Super-phosphate - Constituents; 100g BCR-038 Fly ash from pulverised coal - Trace elements; 5g BCR-039B Pyrex glass -

VIII. Appendix 'G =: 0
'0> VIII. Appendix 'g =: 0.. Structures, systematic, trivial, generic and trade names of protein anabolic steroids ~. Structure Systematic name" Trivial and/or generic name Trade name Manufacturer L.T!'''..tosterQ!.le-deril1ed l!.rote~anabolic steroids 17 fi-Hydroxyandrost- Testosterone Geno-cristaux Gremy H 4-en-3-one Malestrone Kirk, N.Y. cOVo Orquisteron/Frosst (Columbia) Primotest Schering A.G. o 0 Oreton Schering -acetate Aceto-Sterandryl Roussel Aceto-Testoviron Schering A.G. Perandrone A ClBA -propionate Oreton Schering Perandren Ciba-Geigy Sterandryl Roussel Testoviron Schering Anertan Boehringer (M) Enarmon Takeda (Japan) -(3-p-hexyloxiphenyl)- Androdurin Leo, propionate (Helsingborg) -heptanoate (enanthate) Delatestryl Squibb Androtardyl Schering-Sepps Testoenant Geymonod -cycJopentyl propionate Depot testosterone Upjohn cypionate 4-Ch1oro-17 fi-hydroxy- Chlorotestosterone Steranabol Farmitalia androst-4-en-3-one -acetate Macrobin Teikoku ~Ol! Turinabol Jenapharm Turinabol inj. Jenapharm 0\ o 0 IV -capronate Macrobin-depot Teikoku -...J Cl 0\ Appendix (continued) tv 00 Structure Systematic namea Trivial and/or generic name Trade name Manufacturer 17 P-Hydroxy-17-methyl- Methyltestosterone Metandren Ciba androst-4-en-3-one Anertan Boehringer (M) Oreton Schering *OH Android Brown o ~ CH 3 9cx-Fluoro-llp,17P-dihydroxy- Fluoxymesterone Halotestin Upjohn 17-methylandrost-4-en-3-one Ultandren Ciba Androfluorene Midy 0£UOH Fluotestin Roter o ~ Orateston Hoechst CH 3 4,17 P-Dihydroxy-17-methyl- Oxymesterone, Oranabol Farmitalia androst -4-en -3-one 4-Hydroxymethyltestosterone Kabi Lipfa Q5DOIl May & Baker o ~ Sanaboral Ikapharm Theranabol Theraplix OH IIIX,17 P-Dihydroxy-17-methyl- lllX-Hydroxy- etsDoH androst-4-en-3-one methyltestosterone o ~ 17p-Hydroxy-17-methyl- 11-0xomethyltestosterone androst-4-ene-3,11-dione ct)BoH '0>- '0 (1) ::l o ~ 0- >;" Appendix (continued) Structure Systematic name" Trivial and/or generic name Trade name Manufacturer '0;..- '0 (1l CH 3 4-Chloro-17 fJ-hydroxy-17 -methyl- Chloromethyitestosterone Turinabol tab!. -
Code Product Api / Imp Unit Ref
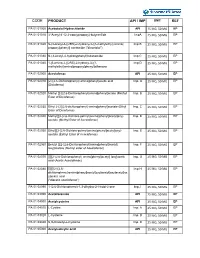
CODE PRODUCT API / IMP UNIT REF PA 01 01000 Acebutolol Hydrochloride API 25 MG, 50 MG EP PA 01 01010 3’-Acetyl-4’-(2,3-epoxypropoxy)-butyranilide ImpA 25 MG, 50 MG EP PA 01 01020 N-[3-acetyl-4-[(2RS)-2-hydroxy-3-[(1-methylethyl) amino] Imp B 25 MG, 50 MG EP propoxy] phenyl] acetamide ("diacetolol") PA 01 01030 N-(3-Acetyl-4-hydroxyphenyl)butanamide Imp C 25 MG, 50 MG EP PA 01 01040 1-[5-amino-2-[(2RS)-2-hydroxy-3-[(1- Imp D 25 MG, 50 MG EP methylethyl)amino]propoxy]phenyl]ethanone PA 01 02000 Aceclofenac API 25 MG, 50 MG EP PA 01 02010 [2-[(2,6-Dichlorophenyl) amino]phenyl]acetic acid Imp. A 25 MG, 50 MG EP (Diclofenac) PA 01 02020 Methyl [2-[(2,6-Dichlorophenyl)amino]phenyl]actate (Methyl Imp. B 25 MG, 50 MG EP Ester of Diclofenac) PA 01 02030 Ethyl 2-(2[(2,6-dichlorophenyl) amino]phenyl]acetate (Ethyl Imp. C 25 MG, 50 MG EP Ester of Diclofenac) PA 01 02040 Methyl[[[2-[2,6-Dichloro-pehnyl)amino)phenyl]acetyl]oxy)- Imp. D 25 MG, 50 MG EP acetate (y(Methyl Ester of Aceclofenac) ) PA 01 02050 Ethyl[[[2-[2,6-Dichloro-pehnyl)amino)phenyl]acetyl]oxy)- Imp. E 25 MG, 50 MG EP acetate (Eethyl Ester of Aceclofenac) PA 01 02060 Benzyl [[[2-(2,6-Dichlorophenyl)amino]phenyl]acetyl] Imp. F 25 MG, 50 MG EP loxy]acetate (Benzyl ester of Aceclofenac) PA 01 02070 [[[[[2-(2,6-Dichlorophenyl) amino]phenyl]acetyl] loxy]acetic Imp. -

Testosterone Art. 31
Příloha I Seznam názvů léčivých přípravků, lékové formy, koncentrace, způsoby podání, a držitelé rozhodnutí o registraci v členských státech 1 Členský Držitel rozhodnutí o INN Smyšlený název Koncentrace Léková forma Způsob podání stát registraci EU/EHP Rakousko Bayer Austria GmbH testosterone undecanoat Nebido 1000 mg/4 ml 1000 mg/4ml Injekční roztok Intramuskulární Herbststrasse 6-10 Injektionslösung podání 1160 Wien Austria Rakousko Eli Lilly GmbH testosterone Axiron 30 mg/1,5 ml 30 mg/1,5ml Gel Kožní podání Kölblgasse 8-10 Lösung zur 1030 Wien Anwendung auf der Austria Haut Rakousko Laboratoires Besins testosterone Testogel 25 mg - Gel 25 mg Gel Kožní podání International SA im Beutel Rue de Bourg lÀbbè 3 75003 Paris France Rakousko Laboratoires Besins testosterone Testogel 50 mg - Gel 50 mg Gel Kožní podání International SA im Beutel Rue de Bourg lÀbbè 3 75003 Paris France Rakousko ProStrakan Ltd. testosterone Tostran 2% - Gel 0.02 Gel Kožní podání Galabank Business Park TD1 1QH Galashiels United Kingdom Rakousko Laboratoires Besins testosterone Androgel 25 mg - Gel 25 mg Gel Kožní podání International SA im Beutel Rue de Bourg lÀbbè 3 75003 Paris France Rakousko Laboratoires Besins testosterone Androgel 50 mg - Gel 50 mg Gel Kožní podání International SA im Beutel Rue de Bourg lÀbbè 3 75003 Paris France 2 Členský Držitel rozhodnutí o INN Smyšlený název Koncentrace Léková forma Způsob podání stát registraci EU/EHP Rakousko N.V. Organon testosterone undecanoat Andriol Testocaps 40 40 mg Měkká tobolka Perorální podání Kloosterstraat 6 mg - Kapseln 5349 AB Oss The Netherlands Belgie Besins Healthcare testosterone Androgel 25 mg Gel Transdermální Avenue Louise 287 podání 1050 Brussels Belgium Belgie Besins Healthcare testosterone Androgel 50 mg Gel Transdermální Avenue Louise 287 podání 1050 Brussels Belgium Belgie Eli Lilly Benelux S.A.-N.V.