Molecular Dynamics Simulation of Ion Mobility. 2. Alkali Metal and Halide Ions Using the SPC/E Model for Water at 25 °C†
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Influence of Water on Conductivity of Aluminium Halide Solutions in Acetonitrile
Influence of water on conductivity of aluminium halide solutions in acetonitrile L. LUX Department of Chemistry, Faculty of Metallurgy, Technical University, CS-043 85 Košice Received 7 September 1981 Accepted for publication 8 February 1982 Equilibria of the substitution and association reactions of AICI3 and AlBr3 with acetonitrile were investigated by measuring the conductivity of solutions at varying concentrations of A1C13, AlBr3, and water in the acetonitrile solution. Both halides behave in acetonitrile in the absence of water as relatively strong electrolytes. It has been revealed that water displaces acetoni trile from the solvate sphere of cation of both halides. As for AlBr3, the bromide ion is also displaced from the coordination sphere of aluminium and replaced by a neutral water molecule, which simultaneously results in an increased number of ionized particles in solution. The hydrate A1X3 -6H20 was identified as final product which separates from the solution by adding water. Изучены равновесия реакций замещения и ассоциации А1С13 и А1Вг3 с ацетонитрилом посредством измерения электропроводности раствора в зависимости от концентрации А1С13, А1Вг3, как и в зависимости от концентрации воды в растворе ацетонитрила. Оба галогенида ведут себя в ацетонитриле как сравнительно сильные электролиты в отсутствии воды. Обнаружено, что вода вытесняет ацетонитрил из сольватной обо лочки катиона у обоих галогенидов. У А1Вг3 доходит и к вытеснению иона брома из координационной сферы алюминия и его замещению нейтраль ной молекулой воды, что одновременно приводит к увеличению числа ионизированных частиц в растворе. Конечным продуктом, выделенным из раствора при добавлении воды был идентифицированный гидрат А1Х3-6Н20. Acetonitrile is a typical aprotic solvent which is widely used in electrochemical research because of its profitable properties, i.e. -

Halides and Halogens. What Do I Need to Know? John Vivari, Nordson EFD
Halides and Halogens. What do I need to know? John Vivari, Nordson EFD Abstract With halogen-containing substances in the public eye due to scrutiny by the European Union and a variety of non- governmental organizations (NGOs) as possible additions to the list of substances banned from electronics, we at EFD have received numerous inquiries from customers asking how this subject will affect them and their processes. Having just overcome the hurdle of RoHS (Restriction of Hazardous Substances), they want to know what halogens and halides are, and what changes they should be prepared for if required to stop using them. Halide-free materials are not new. Some segments of the electronics industry have been sensitive to halides and their significance for decades. This paper will give the reader a working knowledge of halogens and halides. Armed with this education, the reader will be able to make informed decisions when required to use halogen-free materials, either because regulations dictate it or social pressure makes acceptance preferable to resistance. Key Words: halide, halogen, bromine, chlorine, flame retardant, RoHS What are halogens and halides? damage. Brominated flame retardant use is not limited to electronics. It is also in common usage in furniture, At their most basic level, halogens are the electronegative construction materials and textiles. elements in column 17 of the periodic table, including fluorine (F), chlorine, (Cl), bromine (Br), iodine (I) and Other sources of halogens in circuit boards include astatine (At). In electronics fiberglass sizing, epoxy curing agents and accelerators, applications, iodine and resin wetting and de-foaming agents, flux residues, and astatine are rarely if ever contamination from handling. -
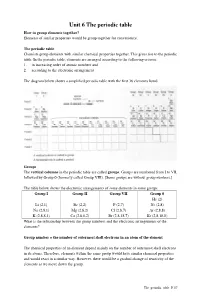
Unit 6 the Periodic Table How to Group Elements Together? Elements of Similar Properties Would Be Group Together for Convenience
Unit 6 The periodic table How to group elements together? Elements of similar properties would be group together for convenience. The periodic table Chemists group elements with similar chemical properties together. This gives rise to the periodic table. In the periodic table, elements are arranged according to the following criteria: 1. in increasing order of atomic numbers and 2. according to the electronic arrangement The diagram below shows a simplified periodic table with the first 36 elements listed. Groups The vertical columns in the periodic table are called groups . Groups are numbered from I to VII, followed by Group 0 (formerly called Group VIII). [Some groups are without group numbers.] The table below shows the electronic arrangements of some elements in some groups. Group I Group II Group VII Group 0 He (2) Li (2,1) Be (2,2) F (2,7) Ne (2,8) Na (2,8,1) Mg (2,8,2) Cl (2,8,7) Ar (2,8,8) K (2,8,8,1) Ca (2,8,8,2) Br (2,8,18,7) Kr (2,8,18,8) What is the relationship between the group numbers and the electronic arrangements of the elements? Group number = the number of outermost shell electrons in an atom of the element The chemical properties of an element depend mainly on the number of outermost shell electrons in its atoms. Therefore, elements within the same group would have similar chemical properties and would react in a similar way. However, there would be a gradual change of reactivity of the elements as we move down the group. -

The Elements.Pdf
A Periodic Table of the Elements at Los Alamos National Laboratory Los Alamos National Laboratory's Chemistry Division Presents Periodic Table of the Elements A Resource for Elementary, Middle School, and High School Students Click an element for more information: Group** Period 1 18 IA VIIIA 1A 8A 1 2 13 14 15 16 17 2 1 H IIA IIIA IVA VA VIAVIIA He 1.008 2A 3A 4A 5A 6A 7A 4.003 3 4 5 6 7 8 9 10 2 Li Be B C N O F Ne 6.941 9.012 10.81 12.01 14.01 16.00 19.00 20.18 11 12 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 3 Na Mg IIIB IVB VB VIB VIIB ------- VIII IB IIB Al Si P S Cl Ar 22.99 24.31 3B 4B 5B 6B 7B ------- 1B 2B 26.98 28.09 30.97 32.07 35.45 39.95 ------- 8 ------- 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 4 K Ca Sc Ti V Cr Mn Fe Co Ni Cu Zn Ga Ge As Se Br Kr 39.10 40.08 44.96 47.88 50.94 52.00 54.94 55.85 58.47 58.69 63.55 65.39 69.72 72.59 74.92 78.96 79.90 83.80 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 5 Rb Sr Y Zr NbMo Tc Ru Rh PdAgCd In Sn Sb Te I Xe 85.47 87.62 88.91 91.22 92.91 95.94 (98) 101.1 102.9 106.4 107.9 112.4 114.8 118.7 121.8 127.6 126.9 131.3 55 56 57 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 6 Cs Ba La* Hf Ta W Re Os Ir Pt AuHg Tl Pb Bi Po At Rn 132.9 137.3 138.9 178.5 180.9 183.9 186.2 190.2 190.2 195.1 197.0 200.5 204.4 207.2 209.0 (210) (210) (222) 87 88 89 104 105 106 107 108 109 110 111 112 114 116 118 7 Fr Ra Ac~RfDb Sg Bh Hs Mt --- --- --- --- --- --- (223) (226) (227) (257) (260) (263) (262) (265) (266) () () () () () () http://pearl1.lanl.gov/periodic/ (1 of 3) [5/17/2001 4:06:20 PM] A Periodic Table of the Elements at Los Alamos National Laboratory 58 59 60 61 62 63 64 65 66 67 68 69 70 71 Lanthanide Series* Ce Pr NdPmSm Eu Gd TbDyHo Er TmYbLu 140.1 140.9 144.2 (147) 150.4 152.0 157.3 158.9 162.5 164.9 167.3 168.9 173.0 175.0 90 91 92 93 94 95 96 97 98 99 100 101 102 103 Actinide Series~ Th Pa U Np Pu AmCmBk Cf Es FmMdNo Lr 232.0 (231) (238) (237) (242) (243) (247) (247) (249) (254) (253) (256) (254) (257) ** Groups are noted by 3 notation conventions. -

“All in the Family” Properties of Halogens SCIENTIFIC Periodic Trends and the Properties of the Elements
“All in the Family” Properties of Halogens SCIENTIFIC Periodic Trends and the Properties of the Elements Introduction In every family there are similarities and differences. The same is true in chemical families as well. Elements that share similar chemical properties are arranged in vertical columns, called groups or families, in the modern periodic table. For example, the Group 17 elements, consisting of fluorine, chlorine, bromine, iodine, and astatine, are called the halogens. What are the similarities and differences in the chemical properties of the halogens and their compounds? Concepts • Halogens • Periodic table • Groups or families • Periodic trends Activity Overview The purpose of this demonstration is to explore the similarities and differences in the chemical properties of the halogens. The reactions of chlorine, bromine, and iodine with sodium chloride, sodium bromide, and sodium iodide will be inves- tigated in order to determine the periodic trend for the reactivity of the halogens. Chlorine, bromine, and iodine will be identified by their unique colors in water and hexane. Materials Bromine water, Br2 in H2O, 10 mL† Sodium thiosulfate solution, 4%, 500 mL§ Chlorine water, Cl2 in H2O, 10 mL† Water, distilled or deionized Iodine solution, I2, 0.04 M, 10 mL Bottles (with caps), square glass, 30-mL, 6 Hexanes, C6H14, 60 mL Graduated cylinders, 10- and 25-mL Sodium bromide solution, NaBr, 0.1 M, 10 mL Marking pencil or pen Sodium chloride solution, NaCl, 0.1 M, 10 mL Periodic table Sodium iodide solution, NaI, 0.1 M, 10 mL †Prepare fresh within one day of use—see the Preparation of Solutions section. -

THE PHYSICAL CHEMISTRY of METALS in THEIR MOLTEN HALIDES THESIS Presented for the Degree of DOCTOR of PHILOSOPHY in the UNIVERSI
THE PHYSICAL CHEMISTRY OF METALS IN THEIR MOLTEN HALIDES THESIS presented for the degree of DOCTOR OF PHILOSOPHY in the UNIVERSITY OF LONDON By Lars-Ingvar Staffansson, Bergsingenjor, (Stockholm). London, December 1959, ABSTRACT The phase diagrams of the systems calcium- calcium chloride, calcium-calcium bromide and calcium- calcium iodide have been established by solubility measurements and differential thermal analysis. Each of the systems was found to have a eutectic a mono- tectic and a large miscibility gap. The eutectics are 3.3, 3.2 and 6.8 mole % calcium respectively, and the corresponding temperatures are 763, 728 and 760°C. The monotectics are at 99.5, 99.6 and 99.7 mole % calcium respectively and the corresponding temperatures are 826, 828 and 831°C. The consolute points are at 1338, 1335 and 1380°C and the corresponding compositions are 62, 64 and 74 mole % calcium. Discrepancies between this and previous work are mainly attributed to composition changes of the phases during quenching. A method has been developed that overcomes this difficulty. The depression of the freezing point of strontium by strontium chloride has also been determined. From the results of this work a model for the mutal solubility of metalS and their molten halides has been suggested. CONTENTS Page CHAPTER 1: INTRODUCTION 1 1.1 General Introduction 1 1.2 Relevant previous work on metal-molten salts. 4 1.2.1 General 4 1.2.2 Alkaline earth metal-metal halide systems. 12 CHAPTER 2: EXPERIMENTAL 24 2.1 Programme 24 2.2 Preparation and purity of materials 25 2.2.1 Calcium 25 2.2.2 Strontium 26 2.2.3 Calcium Chloride 26 2.2.4 Calcium Bromide 28 2.2.5 Calcium Iodide 28 2.2.6 Strontium Chloride 29 2.2.7 Pure Iron 30 2.2.8 Gases 31 2.3 Dry-box. -

Chapter 2 Incandescent Light Bulb
Lamp Contents 1 Lamp (electrical component) 1 1.1 Types ................................................. 1 1.2 Uses other than illumination ...................................... 2 1.3 Lamp circuit symbols ......................................... 2 1.4 See also ................................................ 2 1.5 References ............................................... 2 2 Incandescent light bulb 3 2.1 History ................................................. 3 2.1.1 Early pre-commercial research ................................ 4 2.1.2 Commercialization ...................................... 5 2.2 Tungsten bulbs ............................................. 6 2.3 Efficacy, efficiency, and environmental impact ............................ 8 2.3.1 Cost of lighting ........................................ 9 2.3.2 Measures to ban use ...................................... 9 2.3.3 Efforts to improve efficiency ................................. 9 2.4 Construction .............................................. 10 2.4.1 Gas fill ............................................ 10 2.5 Manufacturing ............................................. 11 2.6 Filament ................................................ 12 2.6.1 Coiled coil filament ...................................... 12 2.6.2 Reducing filament evaporation ................................ 12 2.6.3 Bulb blackening ........................................ 13 2.6.4 Halogen lamps ........................................ 13 2.6.5 Incandescent arc lamps .................................... 14 2.7 Electrical -

Chemistry 101 Experiment 10 PERIODIC PROPERTIES When The
Chemistry 101 Experiment 10 PERIODIC PROPERTIES When the elements in the periodic table are arranged in order of increasing atomic number, it is found that elements can be grouped together according to their similar chemical and physical properties. These groups are represented by the vertical columns in the periodic table. Two groups will be investigated in this experiment, Group 2 (IIA), the alkaline earth metals, and Group 17 (VIIA), the halogens. The Group 2 metals are quite reactive and form ions of a 2+ charge. If the combination of the Group 2 cation and an anion, Z2–, leads to a product which is insoluble in water, mixing aqueous solutions of the ions will lead to a precipitate: 2+ 2– M (aq) + Z (aq) MZ(s) If solubility in water of the compounds of the type MZ is a periodic property, then the relative solubilities should be predictable from the periodic table. For example, we might find a variation in solubility as follows: MgZ > CaZ > SrZ > BaZ In this experiment, the solubilities of the compounds of Mg2+, Ca2+, Sr2+, and Ba2+ will be investigated. Compounds of Be2+ are toxic and Ra is a rare, radioactive element, so their properties will not be investigated, but predictions will be made. The anions that will be used are 2– 2– 2– 2– SO4 , CO3 , C2O4 , and CrO4 . The Group 17 elements, the halogens, are also quite reactive. They all occur as diatomic – molecules, X2, in the elemental state and tend to react by gaining electrons to form anions, X . When a halogen reacts with a metal, the halogen acquires electrons from the metal forming a metal cation and a halide anion, F–, Cl–, Br–, I–, or At–. -

3.1.3 Halogens Fluorine (F 2): Very Pale Yellow Gas
3.1.3 Halogens Fluorine (F 2): very pale yellow gas. It is highly reactive All halogens exist as Chlorine : (Cl 2) greenish, reactive gas, poisonous in high concentrations diatomic molecules Bromine (Br 2) : red liquid, that gives off dense brown/orange poisonous fumes Iodine (I 2) : shiny grey solid sublimes to purple gas. Trend in melting point and boiling point Increase down the group As the molecules become larger they have more electrons and so have larger induced dipole-dipole forces (London forces) between the molecules. As the intermolecular forces get larger more energy has to be put into break theses intemolecular forces. This increases the melting and boiling points Redox reactions and reactivity of halogens and their compounds Electronic configuration. All group 7 elements have the outer shell s 2p5 electron configuration. The will often react by gaining of one electron in redox reactions to form 1 – ions 1. The displacement reactions of halide ions by halogens. A halogen that is more reactive will displace The reactivity of the halogens decreases down the group a halogen that has a lower reactivity from one as the atoms get bigger with more shielding so they less of its compounds easily attract and accept electrons. They therefore form -1 ions less easily down the group know these Chlorine will displace both bromide and iodide ions; bromine will displace iodide ions observations ! Chlorine (aq) Bromine (aq) Iodine (aq) The colour of the solution in the test tube shows which free potassium Very pale green Yellow solution, no Brown solution, halogen is present in solution. -
Method 26 Determination of Hydrogen Halide and Halogen Emissions from Stationary Sources Non-Isokinetic 1-14
Method 26 1/14/2019 While we have taken steps to ensure the accuracy of this Internet version of the document, it is not the official version. To see a complete version including any recent edits, visit: https://www.ecfr.gov/cgi- bin/ECFR?page=browse and search under Title 40, Protection of Environment. METHOD 26—DETERMINATION OF HYDROGEN HALIDE AND HALOGEN EMISSIONS FROM STATIONARY SOURCES NON-ISOKINETIC METHOD 1.0 Scope and Application 1.1 Analytes. Analytes CAS No. Hydrogen Chloride (HCl) 7647-01-0 Hydrogen Bromide (HBr) 10035-10-6 Hydrogen Fluoride (HF) 7664-39-3 Chlorine (Cl2) 7882-50-5 Bromine (Br2) 7726-95-6 1.2 Applicability. This method is applicable for determining emissions of hydrogen halides (HX) (HCl, HBr, and HF) and halogens (X2) (Cl2 and Br2) from stationary sources when specified by the applicable subpart. Sources, such as those controlled by wet scrubbers, that emit acid particulate matter must be sampled using Method 26A. 1.3 Data Quality Objectives. Adherence to the requirements of this method will enhance the quality of the data obtained from air pollutant sampling methods. 2.0 Summary of Method 2.1 An integrated sample is extracted from the source and passed through a prepurged heated probe and filter into dilute sulfuric acid and dilute sodium hydroxide solutions which collect the gaseous hydrogen halides and halogens, respectively. The filter collects particulate matter including halide salts but is not routinely recovered and analyzed. The hydrogen halides are solubilized in the acidic solution and form chloride (Cl−), bromide (Br−), and fluoride (F−) ions. -

Performance Evaluation of a Halogen Acid Gas Analyzer
Performance Evaluation Of A Halogen Acid Gas Analyzer S. H. Hoke U.S. Army Center for Health Promotion and Preventive Medicine Fort Detrick, Frederick, Maryland 21702-5010 ph 301-619-7231 fax 301-619-2569 C. Herud U.S. Army Aberdeen Test Center Aberdeen Proving Ground, Maryland 21005-5059 ph 410-278-8804 fax 410-278-8832 ABSTRACT Real-time monitoring instrumentation has been developed to measure halogen acid gases and their aerosols resulting from the thermal breakdown of halogenated fire suppression agents under live fire testing conditions. This halogen acid gas analyzer is designed to measure HF, HCl, HBr, and HI simultaneously. Linear response is indicated from laboratory generated gases and aerosols of each of the acids from a few mg/m3 to several thousand mg/m3. It has the capability of extending the extraction module up to 2 m from the monitor, thus minimizing instrumentation losses should the test conditions become too hostile. INTRODUCTION Many of the current fire suppression agents, as well as the candidates for replacing these agents, contain halogens. In combat situations the crew of an armored vehicle may not be able to exit the vehicle in case of fire; therefore, when these agents are used to extinguish the fire, large quantities of halogen acid gas and aerosol may be produced, which can be as life threatening in close quarters as the fire itself. In order to accurately assess exposure levels and potential health hazards, support Congressionally mandated live fire tests, and reduce testing time and costs, accurate measurements of these toxic gases need to be made on a real- time basis. -

Electron Attachment to Hydrogen Halide Clusters E
Subscriber access provided by Johns Hopkins Libraries Electron attachment to hydrogen halide clusters E. L. Quitevis, K. H. Bowen, G. W. Liesegang, and D. R. Herschbach J. Phys. Chem., 1983, 87 (12), 2076-2079• DOI: 10.1021/j100235a011 • Publication Date (Web): 01 May 2002 Downloaded from http://pubs.acs.org on May 5, 2009 More About This Article The permalink http://dx.doi.org/10.1021/j100235a011 provides access to: • Links to articles and content related to this article • Copyright permission to reproduce figures and/or text from this article The Journal of Physical Chemistry is published by the American Chemical Society. 1155 Sixteenth Street N.W., Washington, DC 20036 2076 J. Phys. Chem. 1983, 87, 2076-2079 Electron Attachment to Hydrogen Halide Clusters E. L. Oultevls,+ K. H. Bowen,$ 0. W. Liesegang,' and D. R. Herschbach' mpaftment of Chemlstty, Harvard Unlverslty, Cambrkige, Massachusetts 02 138 (Recelved: October 25, 1982; In Fhal Form: February 15, 1983) Negative ion production by endoergic charge transfer from fast alkali atoms to a crossed molecular beam of hydrogen halide clusters, (HX),, has been studied for HF, HC1, HBr, HI, and mixed HF, HBr clusters. Cluster ions corresponding to n up to the order of 10 are readily detected, although the unit mass resolution required for full identification is only feasible for small clusters (masses 1100 amu). Dissociative attachment to form the "solvated" halide or bihalide anions, [X(HX)],-,-, is the predominant process. No evidence is found for any intact (HX),,- ions. Introduction observable products. "Solvated" halide ions, Cl-(HCl), Often a single molecule cannot bind an excess electron, with n = 1-4, have been observed in the multiple collision whereas a weakly bound cluster of the same molecules evironment of drift tubes."J2 Yet results of swarm-beam might be able to do so.