Dynamics of Single Molecule Stokes Shifts: Influence of Conformation and Environment
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

DEVELOPMENT of a RAMAN SPECTROMETER to STUDY SURFACE-ENHANCED RAMAN SCATTERING by Nandita Biswas, Ridhima Chadha, Sudhir Kapoor, Sisir K
BARC/2011/E/003 BARC/2011/E/003 DEVELOPMENT OF A RAMAN SPECTROMETER TO STUDY SURFACE-ENHANCED RAMAN SCATTERING by Nandita Biswas, Ridhima Chadha, Sudhir Kapoor, Sisir K. Sarkar and Tulsi Mukherjee Radiation & Photochemistry Division 2011 BARC/2011/E/003 GOVERNMENT OF INDIA ATOMIC ENERGY COMMISSION BARC/2011/E/003 DEVELOPMENT OF A RAMAN SPECTROMETER TO STUDY SURFACE-ENHANCED RAMAN SCATTERING by Nandita Biswas, Ridhima Chadha, Sudhir Kapoor, Sisir K. Sarkar and Tulsi Mukherjee Radiation & Photochemistry Division BHABHA ATOMIC RESEARCH CENTRE MUMBAI, INDIA 2011 BARC/2011/E/003 BIBLIOGRAPHIC DESCRIPTION SHEET FOR TECHNICAL REPORT (as per IS : 9400 - 1980) 01 Security classification : Unclassified 02 Distribution : External 03 Report status : New 04 Series : BARC External 05 Report type : Technical Report 06 Report No. : BARC/2011/E/003 07 Part No. or Volume No. : 08 Contract No. : 10 Title and subtitle : Development of a Raman spectrometer to study surface-enhanced Raman scattering 11 Collation : 31 p., 15 figs. 13 Project No. : 20 Personal author(s) : Nandita Biswas; Ridhima Chadha; Sudhir Kapoor; Sisir K. Sarkar; Tulsi Mukherjee 21 Affiliation of author(s) : Radiation and Photochemistry Division , Bhabha Atomic Research Centre, Mumbai 22 Corporate author(s) : Bhabha Atomic Research Centre, Mumbai - 400 085 23 Originating unit : Radiation and Photochemistry Division, BARC, Mumbai 24 Sponsor(s) Name : Department of Atomic Energy Type : Government Contd... BARC/2011/E/003 30 Date of submission : January 2011 31 Publication/Issue date : February 2011 40 Publisher/Distributor : Head, Scientific Information Resource Division, Bhabha Atomic Research Centre, Mumbai 42 Form of distribution : Hard copy 50 Language of text : English 51 Language of summary : English, Hindi 52 No. -
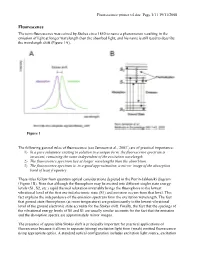
The Term Fluorescence Was Coined by Stokes Circa 1850 to Name A
Fluorescence primer v4.doc Page 1/11 19/11/2008 Fluorescence The term fluorescence was coined by Stokes circa 1850 to name a phenomenon resulting in the emission of light at longer wavelength than the absorbed light, and his name is still used to describe the wavelength shift (Figure 1A). Figure 1 The following general rules of fluorescence (see Jameson et al., 20031) are of practical importance: 1) In a pure substance existing in solution in a unique form, the fluorescence spectrum is invariant, remaining the same independent of the excitation wavelength. 2) The fluorescence spectrum lies at longer wavelengths than the absorbtion. 3) The fluorescence spectrum is, to a good approximation, a mirror image of the absorption band of least frequency These rules follow from quantum optical considerations depicted in the Perrin-Jablonski diagram (Figure 1B). Note that although the fluorophore may be excited into different singlet state energy levels (S1, S2, etc.) rapid thermal relaxation invariably brings the fluorophore to the lowest vibrational level of the first excited electronic state (S1) and emission occurs from that level. This fact explains the independence of the emission spectrum from the excitation wavelength. The fact that ground state fluorophores (at room temperature) are predominantly in the lowest vibrational level of the ground electronic state accounts for the Stokes shift. Finally, the fact that the spacings of the vibrational energy levels of S0 and S1 are usually similar accounts for the fact that the emission and the absorption spectra are approximately mirror images. The presence of appreciable Stokes shift is principally important for practical applications of fluorescence because it allows to separate (strong) excitation light from (weak) emitted fluorescence using appropriate optics. -

Resonant Raman Spectroscopy of Nanotubes
10.1098/rsta.2004.1444 Resonant Raman spectroscopy of nanotubes By Christian Thomsen1, Stephanie Reich2 and Janina Maultzsch1 1Technische Universit¨at Berlin, Hardenbergstraße 36, 10623 Berlin, Germany 2Department of Engineering, University of Cambridge, Trumpington Street, Cambridge CB2 1PZ, UK ([email protected]) Published online 28 September 2004 Single and double resonances in Raman scattering are introduced and six criteria for the observation and identification of double resonances stated. The experimental situation in carbon nanotubes is reviewed in view of these criteria. The evidence for the D mode and the high-energy mode is found to be overwhelming for a double- resonance process to take place, whereas the nature of the radial breathing-mode Raman process remains undecided at this point. Consequences for the application of Raman scattering to the characterization of nanotubes are discussed. Keywords: carbon nanotubes; double resonance; Raman scattering; defects 1. Introduction Raman scattering in carbon nanotubes has developed into a method of choice in the investigation of their physical properties and their characterization. The study of electronic resonances in the Raman spectra, a method which has been used exten- sively, for example, in work on semiconductors (Cardona 1982), gives us a wealth of information about the electronic band structure of a material. This is also true for the Raman work on carbon nanotubes, where resonance studies have moved into the focus of research. Traditional Raman studies of carbon nanotubes focus on the radial breathing mode (RBM), a mode where all atoms vibrate in phase in the radial direction (Dresselhaus et al. 1995). Its frequency, as can easily be shown (Jishi et al. -

Stokes Shift
Stokes shift ticular molecular structure. If a material has a direct bandgap in the range of visible light, the light shining on it is absorbed, causing electrons to become excited to a higher energy state. The electrons remain in the ex- cited state for about 10−8 seconds. This number varies over several orders of magnitude depending on the sam- ple, and is known as the fluorescence lifetime of the sam- ple. After losing a small amount of energy in some way (hence the longer wavelength), the molecule returns to the ground state and energy is emitted. 2 References Absorption and emission spectra of Rhodamine 6G with ~25 nm [1] Gispert, J.R. (2008). Coordination Chemistry. Wiley- Stokes shift VCH. p. 483. ISBN 3-527-31802-X. [2] Albani, J.R. (2004). Structure and Dynamics of Macro- Not to be confused with Stark shift. molecules: Absorption and Fluorescence Studies. Elsevier. p. 58. ISBN 0-444-51449-X. Stokes shift is the difference (in wavelength or frequency [3] Lakowicz, J.R. 1983. Principles of Fluorescence Spec- units) between positions of the band maxima of the troscopy, Plenum Press, New York. ISBN 0-387-31278- absorption and emission spectra (fluorescence and Raman 1. being two examples) of the same electronic transition.[1] [4] Guilbault, G.G. 1990. Practical Fluorescence, Second It is named after Irish physicist George G. Stokes.[2][3][4] Edition, Marcel Dekker, Inc., New York. ISBN 0-8247- When a system (be it a molecule or atom) absorbs a 8350-6. photon, it gains energy and enters an excited state. -

Fiber Amplifiers and Fiber Lasers Based on Stimulated Raman
micromachines Review Fiber Amplifiers and Fiber Lasers Based on Stimulated Raman Scattering: A Review Luigi Sirleto * and Maria Antonietta Ferrara National Research Council (CNR), Institute of Applied Sciences and Intelligent Systems, Via Pietro Castellino 111, 80131 Naples, Italy; [email protected] * Correspondence: [email protected] Received: 10 January 2020; Accepted: 24 February 2020; Published: 26 February 2020 Abstract: Nowadays, in fiber optic communications the growing demand in terms of transmission capacity has been fulfilling the entire spectral band of the erbium-doped fiber amplifiers (EDFAs). This dramatic increase in bandwidth rules out the use of EDFAs, leaving fiber Raman amplifiers (FRAs) as the key devices for future amplification requirements. On the other hand, in the field of high-power fiber lasers, a very attractive option is provided by fiber Raman lasers (FRLs), due to their high output power, high efficiency and broad gain bandwidth, covering almost the entire near-infrared region. This paper reviews the challenges, achievements and perspectives of both fiber Raman amplifier and fiber Raman laser. They are enabling technologies for implementation of high-capacity optical communication systems and for the realization of high power fiber lasers, respectively. Keywords: stimulated raman scattering; fiber optics; amplifiers; lasers; optical communication systems 1. Introduction Optical communication systems require optoelectronic devices, such as sources, detectors and so on, and utilize fiber optics to transmit the light carrying the signals impressed by modulators. Optical fibers are affected by chromatic dispersion, losses, and nonlinearity. Dispersion control is, usually, achieved via fiber geometry and material composition. Losses limit the transmission distance in modern long haul fiber-optic communication systems, so in order to boost a weak signal, optical amplifiers have been developed. -

Theoretical Investigation of Stokes Shifts and Reaction Pathways
Theoretical Investigation of Stokes Shifts and Reaction Pathways by Laken M. Top - ARI IVS Associate of Arts, Science Option Yakima Valley Community College, 2007 Bachelor of Science, Chemistry and Mathematics RES University of Idaho, 2009 SUBMITTED TO THE DEPARTMENT OF CHEMISTRY IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF MASTER OF SCIENCE IN CHEMISTRY AT THE MASSACHUSETTS INSTITUTE OF TECHNOLOGY SEPTEMBER 2012 @ 2012 Massachusetts Institute of Technology. All rights reserved. 1 1-/ Signature of Author: Department of Chemistry July 12, 2012 Certified by: I Troy Van Voorhis ,1 Associate Professor of Chemistry Thesis Supervisor I / Certified by: f -y Jeffrey C. Grossman Associate Professor of Materials Science and Engineering Thesis Supervisor Accepted by: Robert W. Field Professor of Chemistry Chairman, Department Committee on Graduate Theses 1 Theoretical Investigation of Stokes Shifts and Reaction Pathways by Laken M. Top Submitted to the Department of Chemistry on July 12, 2012, in partial fulfillment of the requirements for the degree of Master of Science in Chemistry Abstract Solar thermal fuels and fluorescent solar concentrators provide two ways in which the energy from the sun can be harnessed and stored. While much progress has been made in recent years, there is still much more to learn about the way that these applications work and more efficient materials are needed to make this a feasible source of renewable energy. Theoretical chemistry is a powerful tool which can provide insight into the processes involved and the properties of materials, allowing us to predict substances that might improve the efficiency of these devices. In this work, we explore how the delta self-consistent field method performs for the calculation of Stokes shifts for conjugated dyes. -
Stokes Shift Spectroscopy Highlights Differences of Cancerous and Normal Human Tissues
3360 OPTICS LETTERS / Vol. 37, No. 16 / August 15, 2012 Stokes shift spectroscopy highlights differences of cancerous and normal human tissues Yang Pu, Wuabo Wang, Yuanlong Yang, and R. R. Alfano* Institute for Ultrafast Spectroscopy and Lasers, and Department of Physics, City College of the City University of New York, Convent Avenue at 138th Street, New York, New York 10031, USA *Corresponding author: [email protected] Received May 15, 2012; accepted June 25, 2012; posted June 28, 2012 (Doc. ID 168649); published August 6, 2012 The Stokes shift spectroscopy (S3) offers a simpler and better way to recognize spectral fingerprints of fluorophores in complex mixtures. The efficiency of S3 for cancer detection in human tissue was investigated systematically. The alterations of Stokes shift spectra (S3) between cancerous and normal tissues are due to the changes of key fluor- ophores, e.g., tryptophan and collagen, and can be highlighted using optimized wavelength shift interval. To our knowledge, this is the first time to explicitly disclose how and why S3 is superior in comparison with other conventional spectroscopic techniques. © 2012 Optical Society of America OCIS codes: 170.0170, 170.6510, 300.0300, 300.6170. Fluorescence spectroscopy has been widely investigated These differences may reflect tissue fluorophores’ for diagnosing cancer since the study by Alfano et al. in change during the evolution of cancer. In order to under- the 1980s [1]. The difference between the emission and stand which components mainly contribute to these absorption peaks is known as the Stokes shift interval, changes, S3 of the main fluorophores in breast tissue, Δλss. -

Fluorescent Dyes with Large Stokes Shifts for Super-Resolution Optical Microscopy of Biological Objects: a Review
Home Search Collections Journals About Contact us My IOPscience Fluorescent dyes with large Stokes shifts for super-resolution optical microscopy of biological objects: a review This content has been downloaded from IOPscience. Please scroll down to see the full text. 2015 Methods Appl. Fluoresc. 3 042004 (http://iopscience.iop.org/2050-6120/3/4/042004) View the table of contents for this issue, or go to the journal homepage for more Download details: IP Address: 134.76.223.157 This content was downloaded on 26/11/2015 at 13:35 Please note that terms and conditions apply. IOP Methods and Applications in Fluorescence Methods Appl. Fluoresc. 3 (2015) 042004 doi:10.1088/2050-6120/3/4/042004 Methods Appl. Fluoresc. 3 TOPICAL REVIEW 2015 Fluorescent dyes with large Stokes shifts for super-resolution RECEIVED © 2015 IOP Publishing Ltd 17 April 2015 optical microscopy of biological objects: a review REVISED 28 August 2015 MAFEB2 Maksim V Sednev, Vladimir N Belov and Stefan W Hell ACCEPTED FOR PUBLICATION 2 September 2015 Department of NanoBiophotonics, Max Planck Institute for Biophysical Chemistry, Am Fassberg 11, 37077 Göttingen, Germany 042004 PUBLISHED E-mail: [email protected] 22 October 2015 Keywords: fluorescence, dyes, large Stokes shift, STED microscopy Maksim V Sednev et al Abstract The review deals with commercially available organic dyes possessing large Stokes shifts and their applications as fluorescent labels in optical microscopy based on stimulated emission depletion Printed in the UK (STED). STED microscopy breaks Abbe’s diffraction barrier and provides optical resolution beyond the diffraction limit. STED microscopy is non-invasive and requires photostable fluorescent markers attached to biomolecules or other objects of interest. -

Efficient Raman Amplifiers and Lasers in Optical Fibers and Silicon
Efficient Raman Amplifiers and Lasers in Optical Fibers and Silicon Waveguides: New Concepts Vom Promotionsausschuss der Technischen Universit¨atHamburg-Harburg zur Erlangung des akademischen Grades Doktor-Ingenieur (Dr.-Ing.) genehmigte Dissertation von Michael Krause aus Hamburg 2007 1. Gutachter: Prof. Dr. Ernst Brinkmeyer, TU Hamburg-Harburg 2. Gutachter: Prof. Dr. Klaus Petermann, TU Berlin 3. Gutachter: Prof. Dr. Klaus Sch¨unemann,TU Hamburg-Harburg Tag der m¨undlichen Pr¨ufung:6. Februar 2007 Uniform Resource Name (URN): urn:nbn:de:gbv:830-tubdok-5769 Danksagung Diese Arbeit ist an der Technischen Universit¨at Hamburg-Harburg w¨ahrend meiner T¨atigkeit als wissenschaftlicher Mitarbeiter in der Arbeitsgruppe Optische Kommu- " nikationstechnik\ entstanden. Ganz herzlich danken m¨ochte ich zun¨achst dem Leiter dieser Arbeitsgruppe, Herrn Prof. Dr. Ernst Brinkmeyer, fur¨ die M¨oglichkeit zur Mitar- beit und Promotion, die umfassende Betreuung und die Unterstutzung¨ s¨amtlicher meiner Vorhaben. Zu gr¨oßtem Dank verpflichtet bin ich weiterhin Hagen Renner fur¨ die unz¨ahligen Gelegenheiten zum ¨außerst ergiebigen fachlichen und nicht-fachlichen Diskutieren und Ideenfinden sowie schließlich fur¨ das Korrekturlesen dieser Arbeit. Bei Sven Cierullies m¨ochte ich mich fur¨ die ergebnisreiche Zusammenarbeit auf dem Gebiet der Raman-Faserlaser und die immer angenehme Stimmung im gemeinsamen Buro¨ bedanken. Weiterhin danke ich Raimonda Stanslovaityte, Robert Draheim, Yi Han und Heiko Fimpel, die im Rahmen ihrer Studien- und Diplomarbeiten hilfreiche Beitr¨age zu meiner Arbeit geliefert haben. Auch allen ubrigen¨ Mitarbeitern und Studenten der Arbeitsgruppe sei gedankt fur¨ ihren Beitrag zu einer gelungenen Arbeitsumgebung; J¨org Voigt danke ich fur¨ die Bera- tung bei praktisch-experimentellen Fragen und Frank Knappe fur¨ vielerlei Hilfestellun- gen. -

1 Basic Principles of Fluorescence Spectroscopy
j1 1 Basic Principles of Fluorescence Spectroscopy 1.1 Absorption and Emission of Light As fluorophores play the central role in fluorescence spectroscopy and imaging we will start with an investigation of their manifold interactions with light. A fluorophore is a component that causes a molecule to absorb energy of a specific wavelength and then re-remit energy at a different but equally specific wavelength. The amount and wavelength of the emitted energy depend on both the fluorophore and the chemical environment of the fluorophore. Fluorophores are also denoted as chromophores, historically speaking the part or moiety of a molecule responsible for its color. In addition, the denotation chromophore implies that the molecule absorbs light while fluorophore means that the molecule, likewise,emits light. The umbrella term used in light emission is luminescence, whereas fluorescence denotes allowed transitions with a lifetime in the nanosecond range from higher to lower excited singlet states of molecules. In the following we will try to understand why some compounds are colored and others are not. Therefore, we will take a closer look at the relationship of conjugation to color with fluorescence emission, and investigate the absorption of light at different wavelengths in and near the visible part of the spectrum of various compounds. For example, organic compounds (i.e., hydrocarbons and derivatives) without double or triple bonds absorb light at wavelengths below 160 nm, corre- À sponding to a photon energy of >180 kcal mol 1 (1 cal ¼ 4.184 J), or >7.8 eV (Figure 1.1), that is, significantly higher than the dissociation energy of common carbon-to-carbon single bonds. -

Surface Enhanced Raman Spectroscopy Using a Single Mode Nanophotonic-Plasmonic Platform
Surface Enhanced Raman Spectroscopy Using a Single Mode Nanophotonic-Plasmonic Platform Fred´ eric´ Peyskens,∗,y Ashim Dhakal,y Pol Van Dorpe,z Nicolas Le Thomas,y and Roel Baetsy Photonics Research Group, INTEC-department, Ghent University-imec; Center for Nano-and BioPhotonics, Ghent University, Sint-Pietersnieuwstraat 41, 9000 Ghent, Belgium, and imec, Kapeldreef 75, 3001 Heverlee, Belgium; Department of Physics, KULeuven, Celestijnenlaan 200D, 3001 Leuven, Belgium E-mail: [email protected] Abstract We demonstrate for the first time the generation of Surface Enhanced Raman Spec- troscopy (SERS) signals from integrated bowtie antennas, excited and collected by the fundamental TE-mode of a single mode silicon nitride waveguide. Due to the integrated nature of this particular single mode SERS-probe one can rigorously quantify the com- plete enhancement process. The Stokes power, generated by a 4-Nitrothiophenol-coated antenna and collected into the fundamental TE-mode, exhibits an 8 × 106 enhance- ment compared to the free-space Raman scattering of a 4-Nitrothiophenol molecule. ∗To whom correspondence should be addressed yPhotonics Research Group, INTEC-department, Ghent University-imec; Center for Nano-and BioPho- tonics, Ghent University, Sint-Pietersnieuwstraat 41, 9000 Ghent, Belgium zimec, Kapeldreef 75, 3001 Heverlee, Belgium; Department of Physics, KULeuven, Celestijnenlaan 200D, 3001 Leuven, Belgium 1 Furthermore we present an analytical model which identifies the relevant design pa- rameters and figure of merit for this new SERS-platform. An excellent correspondence is obtained between the theoretically predicted and experimentally observed absolute Raman power. This work paves the way towards a new class of fully integrated lab- on-a-chip systems where the single mode SERS-probe can be combined with other photonic, fluidic or biological functionalities. -

Reorientation-Induced Stokes Shifts Caused by Directional Interactions in Electronic Spectroscopy: Fast Dynamics of Poly(Methyl Methacrylate)
The Journal ARTICLE of Chemical Physics scitation.org/journal/jcp Reorientation-induced Stokes shifts caused by directional interactions in electronic spectroscopy: Fast dynamics of poly(methyl methacrylate) Cite as: J. Chem. Phys. 150, 194201 (2019); doi: 10.1063/1.5094806 Submitted: 5 March 2019 • Accepted: 24 April 2019 • Published Online: 16 May 2019 Joseph E. Thomaz, Patrick L. Kramer, Sebastian M. Fica-Contreras, David J. Hoffman, and Michael D. Fayera) AFFILIATIONS Department of Chemistry, Stanford University, Stanford, California 94305, USA a)[email protected]. Tel.: 650 723-4446. ABSTRACT Dynamic Stokes shift measurements report on structural relaxation, driven by a dipole created in a chromophore by its excitation from the ground electronic state to the S1 state. Here, we demonstrate that it is also possible to have an additional contribution from orientational relax- ation of the Stokes shift chromophore. This effect, called reorientation-induced Stokes shift (RISS), can be observed when the reorientation of the chromophore and the solvent structural relaxation occur on similar time scales. Through a vector interaction, the electronic transition of the chromophore couples to its environment. The orientational diffusive motions of the chromophores will have a slight bias toward reduc- ing the transition energy (red shift) as do the solvent structural diffusive motions. RISS is manifested in the polarization-dependence of the fluorescence Stokes shift using coumarin 153 (C153) in poly(methyl methacrylate) (PMMA). A similar phenomenon, reorientation-induced spectral diffusion (RISD), has been observed and theoretically explicated in the context of two dimensional infrared (2D IR) experiments. Here, we generalize the existing RISD theory to include properties of electronic transitions that generally are not present in vibrational tran- sitions.