1 Basic Principles of Fluorescence Spectroscopy
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Absolute Fluorescence Quantum Yields Using the FL 6500 Fluorescence Spectrometer
APPLICATION NOTE Fluorescence Spectroscopy Authors: Kathryn Lawson-Wood Steve Upstone Kieran Evans PerkinElmer, Inc. Seer Green, UK Absolute Fluorescence Quantum Yields Using the FL 6500 Introduction Fluorescence quantum yield Fluorescence Spectrometer (ΦF) is a characteristic property of a fluorescent species and is denoted as the ratio of the number of photons emitted through fluorescence, to the number of photons absorbed by the fluorophore (Equation 1). ΦF ultimately relates to the efficiency of the pathways leading to emission of fluorescence (Figure 1), providing the probability of the excited state being deactivated by fluorescence rather than by other competing relaxation processes. The magnitude of ΦF is directly related to the intensity of the observed fluorescence.1 Number of photons emitted through fluorescence ΦF = (1) Number of photons absorbed Fluorescence quantum yield can be measured using two methods: the relative method and the absolute method. In comparison with relative quantum yield measurements, the absolute method is applicable to both solid and solution phase measurements. The ΦF can be determined in a single measurement without the need for a reference standard. It is especially useful for samples which absorb and emit in wavelength regions for which there are no reliable quantum yield standards available.2 The importance of ΦF has been shown in several industries, standard. It is especially useful for samples which absorb and including the research, development and evaluation of audio emit in wavelength regions for which there are no reliable visual equipment, electroluminescent materials (OLED/LED) and quantum yield standards available. This method requires an fluorescent probes for biological assays.3 This application integrating sphere which allows the instrument to collect all of demonstrates the use of the PerkinElmer FL 6500 Fluorescence the photons emitted from the sample. -

Dozy-Chaos Mechanics for a Broad Audience
challenges Concept Paper Dozy-Chaos Mechanics for a Broad Audience Vladimir V. Egorov Russian Academy of Sciences, FSRC “Crystallography and Photonics”, Photochemistry Center, 7a Novatorov Street, 119421 Moscow, Russia; [email protected] Received: 29 June 2020; Accepted: 28 July 2020; Published: 31 July 2020 Abstract: A new and universal theoretical approach to the dynamics of the transient state in elementary physico-chemical processes, called dozy-chaos mechanics (Egorov, V.V. Heliyon Physics 2019, 5, e02579), is introduced to a wide general readership. Keywords: theoretical physics; quantum mechanics; molecular quantum transitions; singularity; dozy chaos; dozy-chaos mechanics; electron transfer; condensed matter; applications 1. Introduction In a publication of Petrenko and Stein [1], it is demonstrated that “the feasibility of the theory of extended multiphonon electron transitions” [2–4] “for the description of optical spectra of polymethine dyes and J-aggregates using quantum chemistry”. A recent publication by the same authors [5] “demonstrates the feasibility and applicability of the theory of extended multiphonon electron transitions for the description of nonlinear optical properties of polymethine dyes using quantum chemistry and model calculations”. About twenty years have passed since the first publications on the theory of extended multiphonon electron transitions [2–4], however, apart from the author of this theory and this paper, only Petrenko and Stein managed to use their results constructively, despite their promising character [1,5,6], and despite the fact that for a long time, the new theory has been constantly in the field of view of a sufficiently large number of researchers (see, e.g., [7–31]). The low demand in practice for the new, promising theory is associated not only with its complexity (however, we note that this theory is the simplest theory of all its likely future and more complex modifications [6]), but also with an insufficiently clear presentation of its physical foundations in the author’s pioneering works [2–4]. -

The Green Fluorescent Protein
P1: rpk/plb P2: rpk April 30, 1998 11:6 Annual Reviews AR057-17 Annu. Rev. Biochem. 1998. 67:509–44 Copyright c 1998 by Annual Reviews. All rights reserved THE GREEN FLUORESCENT PROTEIN Roger Y. Tsien Howard Hughes Medical Institute; University of California, San Diego; La Jolla, CA 92093-0647 KEY WORDS: Aequorea, mutants, chromophore, bioluminescence, GFP ABSTRACT In just three years, the green fluorescent protein (GFP) from the jellyfish Aequorea victoria has vaulted from obscurity to become one of the most widely studied and exploited proteins in biochemistry and cell biology. Its amazing ability to generate a highly visible, efficiently emitting internal fluorophore is both intrin- sically fascinating and tremendously valuable. High-resolution crystal structures of GFP offer unprecedented opportunities to understand and manipulate the rela- tion between protein structure and spectroscopic function. GFP has become well established as a marker of gene expression and protein targeting in intact cells and organisms. Mutagenesis and engineering of GFP into chimeric proteins are opening new vistas in physiological indicators, biosensors, and photochemical memories. CONTENTS NATURAL AND SCIENTIFIC HISTORY OF GFP .................................510 Discovery and Major Milestones .............................................510 Occurrence, Relation to Bioluminescence, and Comparison with Other Fluorescent Proteins .....................................511 PRIMARY, SECONDARY, TERTIARY, AND QUATERNARY STRUCTURE ...........512 Primary Sequence from -

Studies on Fluorescence Efficiency and Photodegradation Of
Laser Chemistry, 2002, Vol. 20(2–4), pp. 99–110 Studies on Fluorescence Efficiency and Photodegradation of Rhodamine 6G Doped PMMA Using a Dual Beam Thermal Lens Technique ACHAMMA KURIAN*, NIBU A GEORGE, BINOY PAUL, V. P. N. NAMPOORI and C. P. G. VALLABHAN International School of Photonics, Cochin University of Science & Technology, Cochin 682022, India (Received11September 2002) In this paper we report the use of the dual beam thermal lens technique as a quantitative method to determine absolute fluorescence quantum efficiency and concentration quenching of fluorescence emission from rhodamine 6G doped Poly(methyl methacrylate) (PMMA), prepared with different concentrations of the dye. A comparison of the present data with that reported in the literature indicates that the observed variation of fluorescence quantum yield with respect to the dye concentration follows a similar profile as in the earlier reported observations on rhodamine 6G in solution. The photodegradation of the dye molecules under cw laser excitation is also studied using the present method. Key words: Thermal lens; Fluorescence quantum yield; Photostability; Dye doped polymer 1 INTRODUCTION From the mid 60s, dye lasers have been attractive sources of coherent tunable radiation because of their unique operational flexibility. The salient features of dye lasers are their tunability, with emission from near ultraviolet to the near infrared. High gain, broad spectral bandwidth enabled pulsed and continuous wave operation. Although dyes have been demonstrated to lase in the solid, liquid or gas phases, liquid solutions of dyes in suitable organic * Corresponding author. Catholicate College, Pathanamthitta 689645, India. E-mail: [email protected] 100 A. KURIAN et al. -

DEVELOPMENT of a RAMAN SPECTROMETER to STUDY SURFACE-ENHANCED RAMAN SCATTERING by Nandita Biswas, Ridhima Chadha, Sudhir Kapoor, Sisir K
BARC/2011/E/003 BARC/2011/E/003 DEVELOPMENT OF A RAMAN SPECTROMETER TO STUDY SURFACE-ENHANCED RAMAN SCATTERING by Nandita Biswas, Ridhima Chadha, Sudhir Kapoor, Sisir K. Sarkar and Tulsi Mukherjee Radiation & Photochemistry Division 2011 BARC/2011/E/003 GOVERNMENT OF INDIA ATOMIC ENERGY COMMISSION BARC/2011/E/003 DEVELOPMENT OF A RAMAN SPECTROMETER TO STUDY SURFACE-ENHANCED RAMAN SCATTERING by Nandita Biswas, Ridhima Chadha, Sudhir Kapoor, Sisir K. Sarkar and Tulsi Mukherjee Radiation & Photochemistry Division BHABHA ATOMIC RESEARCH CENTRE MUMBAI, INDIA 2011 BARC/2011/E/003 BIBLIOGRAPHIC DESCRIPTION SHEET FOR TECHNICAL REPORT (as per IS : 9400 - 1980) 01 Security classification : Unclassified 02 Distribution : External 03 Report status : New 04 Series : BARC External 05 Report type : Technical Report 06 Report No. : BARC/2011/E/003 07 Part No. or Volume No. : 08 Contract No. : 10 Title and subtitle : Development of a Raman spectrometer to study surface-enhanced Raman scattering 11 Collation : 31 p., 15 figs. 13 Project No. : 20 Personal author(s) : Nandita Biswas; Ridhima Chadha; Sudhir Kapoor; Sisir K. Sarkar; Tulsi Mukherjee 21 Affiliation of author(s) : Radiation and Photochemistry Division , Bhabha Atomic Research Centre, Mumbai 22 Corporate author(s) : Bhabha Atomic Research Centre, Mumbai - 400 085 23 Originating unit : Radiation and Photochemistry Division, BARC, Mumbai 24 Sponsor(s) Name : Department of Atomic Energy Type : Government Contd... BARC/2011/E/003 30 Date of submission : January 2011 31 Publication/Issue date : February 2011 40 Publisher/Distributor : Head, Scientific Information Resource Division, Bhabha Atomic Research Centre, Mumbai 42 Form of distribution : Hard copy 50 Language of text : English 51 Language of summary : English, Hindi 52 No. -

Microenvironment-Triggered Dual-Activation of a Photosensitizer
www.nature.com/scientificreports OPEN Microenvironment‑triggered dual‑activation of a photosensitizer‑ fuorophore conjugate for tumor specifc imaging and photodynamic therapy Chang Wang1, Shengdan Wang1, Yuan Wang1, Honghai Wu1, Kun Bao2, Rong Sheng1* & Xin Li1* Photodynamic therapy is attracting increasing attention, but how to increase its tumor‑specifcity remains a daunting challenge. Herein we report a theranostic probe (azo‑pDT) that integrates pyropheophorbide α as a photosensitizer and a NIR fuorophore for tumor imaging. The two functionalities are linked with a hypoxic‑sensitive azo group. Under normal conditions, both the phototoxicity of the photosensitizer and the fuorescence of the fuorophore are inhibited. While under hypoxic condition, the reductive cleavage of the azo group will restore both functions, leading to tumor specifc fuorescence imaging and phototoxicity. The results showed that azo‑PDT selectively images BEL‑7402 cells under hypoxia, and simultaneously inhibits BEL‑7402 cell proliferation after near‑infrared irradiation under hypoxia, while little efect on BEL‑7402 cell viability was observed under normoxia. These results confrm the feasibility of our design strategy to improve the tumor‑ targeting ability of photodynamic therapy, and presents azo‑pDT probe as a promising dual functional agent. Cancer is one of the most common causes of death, and more and more therapeutic strategies against this fatal disease have emerged in the past few decades. Among these strategies, photodynamic therapy has attracted much attention1. Tis therapy is based on singlet oxygen produced by photosensitizers under the irradiation with light of a specifc wavelength to damage tumor tissues (Fig. 1a). Since the photo-damaging efect is induced by the interaction between a photosensitizer and light, tumor-specifc therapy may be realized by focusing the light to the tumor site. -
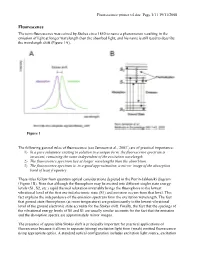
The Term Fluorescence Was Coined by Stokes Circa 1850 to Name A
Fluorescence primer v4.doc Page 1/11 19/11/2008 Fluorescence The term fluorescence was coined by Stokes circa 1850 to name a phenomenon resulting in the emission of light at longer wavelength than the absorbed light, and his name is still used to describe the wavelength shift (Figure 1A). Figure 1 The following general rules of fluorescence (see Jameson et al., 20031) are of practical importance: 1) In a pure substance existing in solution in a unique form, the fluorescence spectrum is invariant, remaining the same independent of the excitation wavelength. 2) The fluorescence spectrum lies at longer wavelengths than the absorbtion. 3) The fluorescence spectrum is, to a good approximation, a mirror image of the absorption band of least frequency These rules follow from quantum optical considerations depicted in the Perrin-Jablonski diagram (Figure 1B). Note that although the fluorophore may be excited into different singlet state energy levels (S1, S2, etc.) rapid thermal relaxation invariably brings the fluorophore to the lowest vibrational level of the first excited electronic state (S1) and emission occurs from that level. This fact explains the independence of the emission spectrum from the excitation wavelength. The fact that ground state fluorophores (at room temperature) are predominantly in the lowest vibrational level of the ground electronic state accounts for the Stokes shift. Finally, the fact that the spacings of the vibrational energy levels of S0 and S1 are usually similar accounts for the fact that the emission and the absorption spectra are approximately mirror images. The presence of appreciable Stokes shift is principally important for practical applications of fluorescence because it allows to separate (strong) excitation light from (weak) emitted fluorescence using appropriate optics. -

Resonant Raman Spectroscopy of Nanotubes
10.1098/rsta.2004.1444 Resonant Raman spectroscopy of nanotubes By Christian Thomsen1, Stephanie Reich2 and Janina Maultzsch1 1Technische Universit¨at Berlin, Hardenbergstraße 36, 10623 Berlin, Germany 2Department of Engineering, University of Cambridge, Trumpington Street, Cambridge CB2 1PZ, UK ([email protected]) Published online 28 September 2004 Single and double resonances in Raman scattering are introduced and six criteria for the observation and identification of double resonances stated. The experimental situation in carbon nanotubes is reviewed in view of these criteria. The evidence for the D mode and the high-energy mode is found to be overwhelming for a double- resonance process to take place, whereas the nature of the radial breathing-mode Raman process remains undecided at this point. Consequences for the application of Raman scattering to the characterization of nanotubes are discussed. Keywords: carbon nanotubes; double resonance; Raman scattering; defects 1. Introduction Raman scattering in carbon nanotubes has developed into a method of choice in the investigation of their physical properties and their characterization. The study of electronic resonances in the Raman spectra, a method which has been used exten- sively, for example, in work on semiconductors (Cardona 1982), gives us a wealth of information about the electronic band structure of a material. This is also true for the Raman work on carbon nanotubes, where resonance studies have moved into the focus of research. Traditional Raman studies of carbon nanotubes focus on the radial breathing mode (RBM), a mode where all atoms vibrate in phase in the radial direction (Dresselhaus et al. 1995). Its frequency, as can easily be shown (Jishi et al. -

Measurement of Fluorescence Quantum Yields on ISS Instrumentation Using Vinci Yevgen Povrozin and Ewald Terpetschnig
TECHNICAL NOTE Measurement of Fluorescence Quantum Yields on ISS Instrumentation Using Vinci Yevgen Povrozin and Ewald Terpetschnig Introduction The quantum yield Q can also be described by the relative rates of the radiative and non-radiative pathways, which deactivate the excited state: kr Q [1 ] kkr nr where and correspond to radiative and non-radiative processes, respectively. In this equation, kr knr knr describes the sum of the rate constants for the various processes that compete with the emission process. These processes include photochemical, and dissociative processes including other, less well characterized changes that result in a return to the ground state with simultaneous dissipation of the energy of the excited state into heat. These latter processes are collectively called non-radiative transitions and two types have been clearly recognized: intersystem crossing and internal conversion. Intersystem crossing related to the radiationless spin inversion of a singlet state (S1) in the excited state into a triplet state (T1). The luminescence quantum yield is also given as the ratio of the number of photons emitted to the number of photons absorbed by the sample: photonsem Q [ 2] photonsabs While measurements of the “absolute” quantum yield do require more sophisticated instrumentation [1], it is easier to determine the “relative” quantum yield of a fluorophore by comparing it to a standard with a known quantum yield. Some of the most common standards are listed in table below: Conditions for Quantum Yield [Q.Y.] Q.Y. Excitation Q.Y. Ref. Standards [nm] [%] Measurements Cy3 4 PBS 540 2 Cy5 27 PBS 620 2 Cresyl Violet 53 Methanol 580 3 1 ISS TECHNICAL NOTE Conditions for Quantum Yield [Q.Y.] Q.Y. -

Fluorescent Protein-Based Tools for Neuroscience
!1 !2 Fluorescent protein-based tools Outline for neuroscience An animatd primer on biosensor development Fluorescent proteins (FPs) Robert E. Campbell Department of Chemistry Other fluorophore technologies Single FP-based biosensors Imaging Structure & Function in the Nervous System Cold Spring Harbor, July 31, 2019. Lots of structural Lots of structural information information & Transmitted light Fluorescence microscopy of fluorescent color microscopy of live cells live cells No molecular provides molecular information information (more colors = more information) !5 !6 Fluorescence microscopy requires fluorophores Non-natural fluorophores for protein labelling Trends Bioch. Sci., 1984, 9, 88-91. O O O N O N 495 nm 519 nm 557 nm 576 nm - - CO2 CO2 S O O O N O N C N N C S ϕ = quantum yield - - ε = extinction coefficient CO2 CO2 ϕ Brightness ~ * ε S C i.e., for fluorescein N Proteins of interest ϕ = 0.92 N Fluorescein Tetramethylrhodamine ε = 73,000 M-1cm-1 C S (FITC) (TRITC) A non-natural fluorophore must be chemically linked to Non-natural fluorophores made by chemical synthesis a protein of interest… !7 !8 Getting non-natural fluorophores into a cell Some sea creatures make natural fluorophores Trends Bioch. Sci., 1984, 9, 88-91. O O O N O N Bioluminescent - Fluorescent CO2 CO -- S 2 S C N NHN C H S S Chemically labeled proteins of interest Microinjection with micropipet O O O N O N Fluorescent - CO2 CO - S 2 N NH H S …and then manually injected into a cell Some natural fluorophores are genetically encoded proteins http://www.luminescentlabs.org/and can be transplanted into cells as DNA! 228 OSAMU SHIMOMURA, FRANK H. -

Photoremovable Protecting Groups Used for the Caging of Biomolecules
1 1 Photoremovable Protecting Groups Used for the Caging of Biomolecules 1.1 2-Nitrobenzyl and 7-Nitroindoline Derivatives John E.T. Corrie 1.1.1 Introduction 1.1.1.1 Preamble and Scope of the Review This chapter covers developments with 2-nitrobenzyl (and substituted variants) and 7-nitroindoline caging groups over the decade from 1993, when the author last reviewed the topic [1]. Other reviews covered parts of the field at a similar date [2, 3], and more recent coverage is also available [4–6]. This chapter is not an exhaustive review of every instance of the subject cages, and its principal focus is on the chemistry of synthesis and photocleavage. Applications of indi- vidual compounds are only briefly discussed, usually when needed to put the work into context. The balance between the two cage types is heavily slanted to- ward the 2-nitrobenzyls, since work with 7-nitroindoline cages dates essentially from 1999 (see Section 1.1.3.2), while the 2-nitrobenzyl type has been in use for 25 years, from the introduction of caged ATP 1 (Scheme 1.1.1) by Kaplan and co-workers [7] in 1978. Scheme 1.1.1 Overall photolysis reaction of NPE-caged ATP 1. Dynamic Studies in Biology. Edited by M. Goeldner, R. Givens Copyright © 2005 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim ISBN: 3-527-30783-4 2 1 Photoremovable Protecting Groups Used for the Caging of Biomolecules 1.1.1.2 Historical Perspective The pioneering work of Kaplan et al. [7], although preceded by other examples of 2-nitrobenzyl photolysis in synthetic organic chemistry, was the first to apply this to a biological problem, the erythrocytic Na:K ion pump. -

Two-Photon Excitation Fluorescence Microscopy
P1: FhN/ftt P2: FhN July 10, 2000 11:18 Annual Reviews AR106-15 Annu. Rev. Biomed. Eng. 2000. 02:399–429 Copyright c 2000 by Annual Reviews. All rights reserved TWO-PHOTON EXCITATION FLUORESCENCE MICROSCOPY PeterT.C.So1,ChenY.Dong1, Barry R. Masters2, and Keith M. Berland3 1Department of Mechanical Engineering, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139; e-mail: [email protected] 2Department of Ophthalmology, University of Bern, Bern, Switzerland 3Department of Physics, Emory University, Atlanta, Georgia 30322 Key Words multiphoton, fluorescence spectroscopy, single molecule, functional imaging, tissue imaging ■ Abstract Two-photon fluorescence microscopy is one of the most important re- cent inventions in biological imaging. This technology enables noninvasive study of biological specimens in three dimensions with submicrometer resolution. Two-photon excitation of fluorophores results from the simultaneous absorption of two photons. This excitation process has a number of unique advantages, such as reduced specimen photodamage and enhanced penetration depth. It also produces higher-contrast im- ages and is a novel method to trigger localized photochemical reactions. Two-photon microscopy continues to find an increasing number of applications in biology and medicine. CONTENTS INTRODUCTION ................................................ 400 HISTORICAL REVIEW OF TWO-PHOTON MICROSCOPY TECHNOLOGY ...401 BASIC PRINCIPLES OF TWO-PHOTON MICROSCOPY ..................402 Physical Basis for Two-Photon Excitation ............................