Understanding Raman Spectroscopy Principles and Theory Basic Raman Instrumentation
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Comparison of Ultraviolet and Visible Raman Spectra of Supported Metal Oxide Catalysts
8600 J. Phys. Chem. B 2001, 105, 8600-8606 A Comparison of Ultraviolet and Visible Raman Spectra of Supported Metal Oxide Catalysts Yek Tann Chua,† Peter C. Stair,*,† and Israel E. Wachs*,‡ Department of Chemistry, Center for Catalysis and Surface Science and Institute of EnVironmental Catalysis, Northwestern UniVersity, EVanston, Illinois 60208, and Zettlemoyer Center for Surface Studies and Department of Chemical Engineering, Lehigh UniVersity, Bethlehem, PennsylVania 18015 ReceiVed: April 11, 2001 The recent emergence of ultraviolet-wavelength-excited Raman spectroscopy as a tool for catalyst characterization has motivated the question of how UV Raman spectra compare to visible-wavelength-excited Raman spectra on the same catalyst system. Measurements of Raman spectra from five supported metal oxide systems (Al2O3-supported Cr2O3,V2O5, and MoO3 as well as TiO2-supported MoO3 and Re2O7), using visible (514.5 nm) and ultraviolet (244 nm) wavelength excitation have been compared to determine the similarities and differences in Raman spectra produced at the two wavelengths. The samples were in the form of self-supporting disks. Spectra from the oxides, both hydrated as a result of contact with ambient air and dehydrated as a result of calcination or laser-induced heating, were recorded. A combination of sample spinning and translation to produce a spiral pattern of laser beam exposure to the catalyst disk was found to be most effective in minimizing dehydration caused by laser-induced heating. Strong absorption by the samples in the ultraviolet significantly reduced the number of scatterers contributing to the Raman spectrum while producing only modest increases in the Raman scattering cross section due to resonance enhancement. -

Atomic and Molecular Laser-Induced Breakdown Spectroscopy of Selected Pharmaceuticals
Article Atomic and Molecular Laser-Induced Breakdown Spectroscopy of Selected Pharmaceuticals Pravin Kumar Tiwari 1,2, Nilesh Kumar Rai 3, Rohit Kumar 3, Christian G. Parigger 4 and Awadhesh Kumar Rai 2,* 1 Institute for Plasma Research, Gandhinagar, Gujarat-382428, India 2 Laser Spectroscopy Research Laboratory, Department of Physics, University of Allahabad, Prayagraj-211002, India 3 CMP Degree College, Department of Physics, University of Allahabad, Pragyagraj-211002, India 4 Physics and Astronomy Department, University of Tennessee, University of Tennessee Space Institute, Center for Laser Applications, 411 B.H. Goethert Parkway, Tullahoma, TN 37388-9700, USA * Correspondence: [email protected]; Tel.: +91-532-2460993 Received: 10 June 2019; Accepted: 10 July 2019; Published: 19 July 2019 Abstract: Laser-induced breakdown spectroscopy (LIBS) of pharmaceutical drugs that contain paracetamol was investigated in air and argon atmospheres. The characteristic neutral and ionic spectral lines of various elements and molecular signatures of CN violet and C2 Swan band systems were observed. The relative hardness of all drug samples was measured as well. Principal component analysis, a multivariate method, was applied in the data analysis for demarcation purposes of the drug samples. The CN violet and C2 Swan spectral radiances were investigated for evaluation of a possible correlation of the chemical and molecular structures of the pharmaceuticals. Complementary Raman and Fourier-transform-infrared spectroscopies were used to record the molecular spectra of the drug samples. The application of the above techniques for drug screening are important for the identification and mitigation of drugs that contain additives that may cause adverse side-effects. Keywords: paracetamol; laser-induced breakdown spectroscopy; cyanide; carbon swan bands; principal component analysis; Raman spectroscopy; Fourier-transform-infrared spectroscopy 1. -

DEVELOPMENT of a RAMAN SPECTROMETER to STUDY SURFACE-ENHANCED RAMAN SCATTERING by Nandita Biswas, Ridhima Chadha, Sudhir Kapoor, Sisir K
BARC/2011/E/003 BARC/2011/E/003 DEVELOPMENT OF A RAMAN SPECTROMETER TO STUDY SURFACE-ENHANCED RAMAN SCATTERING by Nandita Biswas, Ridhima Chadha, Sudhir Kapoor, Sisir K. Sarkar and Tulsi Mukherjee Radiation & Photochemistry Division 2011 BARC/2011/E/003 GOVERNMENT OF INDIA ATOMIC ENERGY COMMISSION BARC/2011/E/003 DEVELOPMENT OF A RAMAN SPECTROMETER TO STUDY SURFACE-ENHANCED RAMAN SCATTERING by Nandita Biswas, Ridhima Chadha, Sudhir Kapoor, Sisir K. Sarkar and Tulsi Mukherjee Radiation & Photochemistry Division BHABHA ATOMIC RESEARCH CENTRE MUMBAI, INDIA 2011 BARC/2011/E/003 BIBLIOGRAPHIC DESCRIPTION SHEET FOR TECHNICAL REPORT (as per IS : 9400 - 1980) 01 Security classification : Unclassified 02 Distribution : External 03 Report status : New 04 Series : BARC External 05 Report type : Technical Report 06 Report No. : BARC/2011/E/003 07 Part No. or Volume No. : 08 Contract No. : 10 Title and subtitle : Development of a Raman spectrometer to study surface-enhanced Raman scattering 11 Collation : 31 p., 15 figs. 13 Project No. : 20 Personal author(s) : Nandita Biswas; Ridhima Chadha; Sudhir Kapoor; Sisir K. Sarkar; Tulsi Mukherjee 21 Affiliation of author(s) : Radiation and Photochemistry Division , Bhabha Atomic Research Centre, Mumbai 22 Corporate author(s) : Bhabha Atomic Research Centre, Mumbai - 400 085 23 Originating unit : Radiation and Photochemistry Division, BARC, Mumbai 24 Sponsor(s) Name : Department of Atomic Energy Type : Government Contd... BARC/2011/E/003 30 Date of submission : January 2011 31 Publication/Issue date : February 2011 40 Publisher/Distributor : Head, Scientific Information Resource Division, Bhabha Atomic Research Centre, Mumbai 42 Form of distribution : Hard copy 50 Language of text : English 51 Language of summary : English, Hindi 52 No. -

Laser Raman Spectroscopy As a Technique for Identification Of
ARTICLE IN PRESS CHEMGE-15589; No of Pages 13 Chemical Geology xxx (2008) xxx–xxx Contents lists available at ScienceDirect Chemical Geology journal homepage: www.elsevier.com/locate/chemgeo Laser Raman spectroscopy as a technique for identification of seafloor hydrothermal and cold seep minerals Sheri N. White ⁎ Department of Applied Ocean Physics and Engineering, Woods Hole Oceanographic Institution, Woods Hole, MA 02536, USA article info abstract Article history: In situ sensors capable of real-time measurements and analyses in the deep ocean are necessary to fulfill the Received 8 August 2008 potential created by the development of autonomous, deep-sea platforms such as autonomous and remotely Received in revised form 8 November 2008 operated vehicles, and cabled observatories. Laser Raman spectroscopy (a type of vibrational spectroscopy) is an Accepted 10 November 2008 optical technique that is capable of in situ molecular identification of minerals in the deep ocean. The goals of this Available online xxxx work are to determine the characteristic spectral bands and relative Raman scattering strength of hydrothermally- Editor: R.L. Rudnick and cold seep-relevant minerals, and to determine how the quality of the spectra are affected by changes in excitation wavelength and sampling optics. The information learned from this work will lead to the development Keywords: of new, smaller sea-going Raman instruments that are optimized to analyze minerals in the deep ocean. Raman spectroscopy Many minerals of interest at seafloor hydrothermal and cold seep sites are Raman active, such as elemental sulfur, Mineralogy carbonates, sulfates and sulfides. Elemental S8 sulfur is a strong Raman scatterer with dominant bands at ∼219 and Hydrothermal vents 472 Δcm−1. -
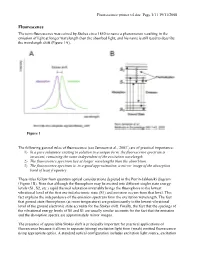
The Term Fluorescence Was Coined by Stokes Circa 1850 to Name A
Fluorescence primer v4.doc Page 1/11 19/11/2008 Fluorescence The term fluorescence was coined by Stokes circa 1850 to name a phenomenon resulting in the emission of light at longer wavelength than the absorbed light, and his name is still used to describe the wavelength shift (Figure 1A). Figure 1 The following general rules of fluorescence (see Jameson et al., 20031) are of practical importance: 1) In a pure substance existing in solution in a unique form, the fluorescence spectrum is invariant, remaining the same independent of the excitation wavelength. 2) The fluorescence spectrum lies at longer wavelengths than the absorbtion. 3) The fluorescence spectrum is, to a good approximation, a mirror image of the absorption band of least frequency These rules follow from quantum optical considerations depicted in the Perrin-Jablonski diagram (Figure 1B). Note that although the fluorophore may be excited into different singlet state energy levels (S1, S2, etc.) rapid thermal relaxation invariably brings the fluorophore to the lowest vibrational level of the first excited electronic state (S1) and emission occurs from that level. This fact explains the independence of the emission spectrum from the excitation wavelength. The fact that ground state fluorophores (at room temperature) are predominantly in the lowest vibrational level of the ground electronic state accounts for the Stokes shift. Finally, the fact that the spacings of the vibrational energy levels of S0 and S1 are usually similar accounts for the fact that the emission and the absorption spectra are approximately mirror images. The presence of appreciable Stokes shift is principally important for practical applications of fluorescence because it allows to separate (strong) excitation light from (weak) emitted fluorescence using appropriate optics. -

Accessing Excited State Molecular Vibrations by Femtosecond Stimulated Raman Spectroscopy
Accessing Excited State Molecular Vibrations by Femtosecond Stimulated Raman Spectroscopy Giovanni Batignani,y Carino Ferrante,y,z and Tullio Scopigno∗,y,z yDipartimento di Fisica, Universitá di Roma “La Sapienza", Roma, I-00185, Italy z Istituto Italiano di Tecnologia, Center for Life Nano Science @Sapienza, Roma, I-00161, Italy E-mail: [email protected] arXiv:2010.05029v1 [physics.optics] 10 Oct 2020 1 Abstract Excited-state vibrations are crucial for determining photophysical and photochem- ical properties of molecular compounds. Stimulated Raman scattering can coherently stimulate and probe molecular vibrations with optical pulses, but it is generally re- stricted to ground state properties. Working in resonance conditions, indeed, enables cross-section enhancement and selective excitation to a targeted electronic level, but is hampered by an increased signal complexity due to the presence of overlapping spectral contributions. Here, we show how detailed information on ground and excited state vi- brations can be disentangled, by exploiting the relative time delay between Raman and probe pulses to control the excited state population, combined with a diagrammatic formalism to dissect the pathways concurring to the signal generation. The proposed method is then exploited to elucidate the vibrational properties of ground and excited electronic states in the paradigmatic case of Cresyl Violet. We anticipate that the presented approach holds the potential for selective mapping the reaction coordinates pertaining to transient electronic stages implied in photo-active compounds. Graphical TOC Entry 2 Raman spectroscopy is a powerful tool to access the vibrational fingerprints of molecules or solid state compounds and it can be used to extract structural and dynamical information of the samples under investigation. -

Resonant Raman Spectroscopy of Nanotubes
10.1098/rsta.2004.1444 Resonant Raman spectroscopy of nanotubes By Christian Thomsen1, Stephanie Reich2 and Janina Maultzsch1 1Technische Universit¨at Berlin, Hardenbergstraße 36, 10623 Berlin, Germany 2Department of Engineering, University of Cambridge, Trumpington Street, Cambridge CB2 1PZ, UK ([email protected]) Published online 28 September 2004 Single and double resonances in Raman scattering are introduced and six criteria for the observation and identification of double resonances stated. The experimental situation in carbon nanotubes is reviewed in view of these criteria. The evidence for the D mode and the high-energy mode is found to be overwhelming for a double- resonance process to take place, whereas the nature of the radial breathing-mode Raman process remains undecided at this point. Consequences for the application of Raman scattering to the characterization of nanotubes are discussed. Keywords: carbon nanotubes; double resonance; Raman scattering; defects 1. Introduction Raman scattering in carbon nanotubes has developed into a method of choice in the investigation of their physical properties and their characterization. The study of electronic resonances in the Raman spectra, a method which has been used exten- sively, for example, in work on semiconductors (Cardona 1982), gives us a wealth of information about the electronic band structure of a material. This is also true for the Raman work on carbon nanotubes, where resonance studies have moved into the focus of research. Traditional Raman studies of carbon nanotubes focus on the radial breathing mode (RBM), a mode where all atoms vibrate in phase in the radial direction (Dresselhaus et al. 1995). Its frequency, as can easily be shown (Jishi et al. -

Absorption Spectroscopy and Imaging from the Visible Through Mid-Infrared with 20 Nm Resolution
Absorption spectroscopy and imaging from the visible through mid-infrared with 20 nm resolution. Aaron M. Katzenmeyer,1 Glenn Holland,1 Kevin Kjoller2 and Andrea Centrone1* 1Center for Nanoscale Science and Technology, National Institute of Standards and Technology, 100 Bureau Drive, Gaithersburg, Maryland 20899, United States. *E-mail: [email protected] 2Anasys Instruments, Inc., 325 Chapala Street, Santa Barbara, California 93101, United States. Abstract Absorption spectroscopy and mapping from visible through mid-IR wavelengths has been achieved with spatial resolution exceeding the limit imposed by diffraction, via the photothermal induced resonance technique. Correlated vibrational (chemical), and electronic properties are obtained simultaneously with topography with a wavelength-independent resolution of ≈ 20 nm using a single laboratory-scale instrument. This marks the highest resolution reported for PTIR, as determined by comparing height and PTIR images, and its first extension to near-IR and visible wavelengths. Light-matter interaction enables scientists to characterize materials and biological samples across a large range of energies. Visible (VIS) and near-infrared (NIR) light (from 400 nm to 2.5 µm) probes electronic transitions in materials [1], providing information regarding band gap, defects, and energy transfer which is crucial for the semiconductor and optoelectronic industries and for understanding processes such as photosynthesis. Mid-infrared (mid-IR) light (from 2.5 µm to 15 µm) probes vibrational transitions and provides rich chemical and structural information enabling materials identification [2]. Spectral maps can be obtained by coupling a light source and a spectrometer with an optical microscope [3]. However, light diffraction limits the lateral resolution achievable with conventional microscopic techniques to approximately half the wavelength of light [4]. -

Raman Scattering and Fluorescence
Fluorescence 01 Raman Scattering and Fluorescence Introduction The existence of such virtual states also explains why the non-resonance Raman effect Raman scattering and Fluorescence emission does not depend on the wavelength of the are two competing phenomena, which have excitation, since no real states are involved in similar origins. Generally, a laser photon this interaction mechanism. In fact, the Raman bounces off a molecule and looses a certain spectrum generally does not depend on the amount of energy that allows the molecule to laser excitation. vibrate (Stokes process). The scattered photon is therefore less energetic and the associated However, when the energy of the excitation light exhibits a frequency shift. The various photon gets close to the transition energy frequency shifts associated with different between two electronic states, one then deals molecular vibrations give rise to a spectrum, with resonance Raman or resonance that is characteristic of a specific compound. fluorescence (fig.1, case (d)). The basic difference between these two processes is In contrast, fluorescence or luminescence related to the time scales involved, as well as emission follows an absorption process. For a with the nature of the so-called intermediate better understanding, one can refer to the states. In contrast with resonant fluorescent, diagram below. relaxed fluorescence results from the emission of a photon from the lowest vibrational level of an excited electronic state, following a direct absorption of the photon and relaxation of the molecule from its vibrationally excited level of the electronic state back to the lowest vibrational level of the electronic state. A fluorescence process typically requires more than 10-9 s. -

Nitrogen-15 Magnetic Resonance Spectroscopy, I
VOL. 51, 1964 CHEMISTRY: LAMBERT, BINSCH, AND ROBERTS 735 11 Felsenfeld, G., G. Sandeen, and P. von Hippel, these PROCEEDINGS, 50, 644 (1963). 12Bollum, F. J., J. Cell. Comp. Physiol., 62 (Suppi. 1), 61 (1963); Von Borstel, R. C., D. M. Prescott, and F. J. Bollum, J. Cell Biol., 19, 72A (1963). 13 Huang, R. C., and J. Bonner, these PROCEEDINGS, 48, 216 (1962); Allfrey, V. G., V. C. Littau, and A. E. Mirsky, these PROCEEDINGS, 49, 414 (1963). 14 Baxill, G. W., and J. St. L. Philpot, Biochim. Biophys. Acta, 76, 223 (1963). NITROGEN-15 MAGNETIC RESONANCE SPECTROSCOPY, I. CHEMICAL SHIFTS* BY JOSEPH B. LAMBERT, GERHARD BINSCH, AND JOHN D. ROBERTS GATES AND CRELLIN LABORATORIES OF CHEMISTRY, t CALIFORNIA INSTITUTE OF TECHNOLOGY Communicated March 23, 1964 Except for the original determination' of nuclear moments, nitrogen magnetic resonance spectroscopy has been limited to the isotope of mass number 14. Al- though N14 is an abundant isotope, it possesses an electric quadrupole moment, which seriously broadens the resonances of nitrogen in all but the most sym- metrical of environments.2 Consequently, nitrogen n.m.r. spectroscopy has seen only limited use in the determination of organic structure. It might be expected that N15, which has a spin of 1/2 and no quadrupole moment, would be very useful, but the low natural abundance (0.36%) and the inherently low signal intensity (1.04 X 10-3 that of H' at constant field) have thus far precluded utilization of N15 in n.m.r. spectroscopy'3 Resonance signals from N'5 have now been obtained from a series of N"5-enriched (30-99%) compounds with a Varian model 4300B spectrometer operated at 6.08 Mc/sec and 14,100 gauss. -

2. Molecular Stucture/Basic Spectroscopy the Electromagnetic Spectrum
2. Molecular stucture/Basic spectroscopy The electromagnetic spectrum Spectral region fooatocadr atomic and molecular spectroscopy E. Hecht (2nd Ed.) Optics, Addison-Wesley Publishing Company,1987 Per-Erik Bengtsson Spectral regions Mo lecu lar spec troscopy o ften dea ls w ith ra dia tion in the ultraviolet (UV), visible, and infrared (IR) spectltral reg ions. • The visible region is from 400 nm – 700 nm • The ultraviolet region is below 400 nm • The infrared region is above 700 nm. 400 nm 500 nm 600 nm 700 nm Spectroscopy: That part of science which uses emission and/or absorption of radiation to deduce atomic/molecular properties Per-Erik Bengtsson Some basics about spectroscopy E = Energy difference = c /c h = Planck's constant, 6.63 10-34 Js ergy nn = Frequency E hn = h/hc /l E = h = hc / c = Velocity of light, 3.0 108 m/s = Wavelength 0 Often the wave number, , is used to express energy. The unit is cm-1. = E / hc = 1/ Example The energy difference between two states in the OH-molecule is 35714 cm-1. Which wavelength is needed to excite the molecule? Answer = 1/ =35714 cm -1 = 1/ = 280 nm. Other ways of expressing this energy: E = hc/ = 656.5 10-19 J E / h = c/ = 9.7 1014 Hz Per-Erik Bengtsson Species in combustion Combustion involves a large number of species Atoms oxygen (O), hydrogen (H), etc. formed by dissociation at high temperatures Diatomic molecules nitrogen (N2), oxygen (O2) carbon monoxide (CO), hydrogen (H2) nitr icoxide (NO), hy droxy l (OH), CH, e tc. -

Absorption Spectroscopy
World Bank & Government of The Netherlands funded Training module # WQ - 34 Absorption Spectroscopy New Delhi, February 2000 CSMRS Building, 4th Floor, Olof Palme Marg, Hauz Khas, DHV Consultants BV & DELFT HYDRAULICS New Delhi – 11 00 16 India Tel: 68 61 681 / 84 Fax: (+ 91 11) 68 61 685 with E-Mail: [email protected] HALCROW, TAHAL, CES, ORG & JPS Table of contents Page 1 Module context 2 2 Module profile 3 3 Session plan 4 4 Overhead/flipchart master 5 5 Evaluation sheets 22 6 Handout 24 7 Additional handout 29 8 Main text 31 Hydrology Project Training Module File: “ 34 Absorption Spectroscopy.doc” Version 06/11/02 Page 1 1. Module context This module introduces the principles of absorption spectroscopy and its applications in chemical analyses. Other related modules are listed below. While designing a training course, the relationship between this module and the others, would be maintained by keeping them close together in the syllabus and place them in a logical sequence. The actual selection of the topics and the depth of training would, of course, depend on the training needs of the participants, i.e. their knowledge level and skills performance upon the start of the course. No. Module title Code Objectives 1. Basic chemistry concepts WQ - 02 • Convert units from one to another • Discuss the basic concepts of quantitative chemistry • Report analytical results with the correct number of significant digits. 2. Basic aquatic chemistry WQ - 24 • Understand equilibrium chemistry and concepts ionisation constants. • Understand basis of pH and buffers • Calculate different types of alkalinity.