Chem 51C Chapter 20 Notes
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

De Novo Biosynthesis of Terminal Alkyne-Labeled Natural Products
De novo biosynthesis of terminal alkyne-labeled natural products Xuejun Zhu1,2, Joyce Liu2,3, Wenjun Zhang1,2,4* 1Department of Chemical and Biomolecular Engineering, 2Energy Biosciences Institute, 3Department of Bioengineering, University of California, Berkeley, CA 94720, USA. 4Physical Biosciences Division, Lawrence Berkeley National Laboratory, Berkeley, CA 94720, USA. *e-mail:[email protected] 1 Abstract: The terminal alkyne is a functionality widely used in organic synthesis, pharmaceutical science, material science, and bioorthogonal chemistry. This functionality is also found in acetylenic natural products, but the underlying biosynthetic pathways for its formation are not well understood. Here we report the characterization of the first carrier protein- dependent terminal alkyne biosynthetic machinery in microbes. We further demonstrate that this enzymatic machinery can be exploited for the in situ generation and incorporation of terminal alkynes into two natural product scaffolds in E. coli. These results highlight the prospect for tagging major classes of natural products, including polyketides and polyketide/non-ribosomal peptide hybrids, using biosynthetic pathway engineering. 2 Natural products are important small molecules widely used as drugs, pesticides, herbicides, and biological probes. Tagging natural products with a unique chemical handle enables the visualization, enrichment, quantification, and mode of action study of natural products through bioorthogonal chemistry1-4. One prevalent bioorthogonal reaction is -

Modern-Reduction-Methods.Pdf
Modern Reduction Methods Edited by Pher G. Andersson and Ian J. Munslow Related Titles Yamamoto, H., Ishihara, K. (eds.) Torii, S. Acid Catalysis in Modern Electroorganic Reduction Organic Synthesis Synthesis 2008 2006 ISBN: 978-3-527-31724-0 ISBN: 978-3-527-31539-0 Roberts, S. M. de Meijere, A., Diederich, F. (eds.) Catalysts for Fine Chemical Metal-Catalyzed Cross- Synthesis V 5 – Regio and Coupling Reactions Stereo-Controlled Oxidations 2004 and Reductions ISBN: 978-3-527-30518-6 2007 Online Book Wiley Interscience Bäckvall, J.-E. (ed.) ISBN: 978-0-470-09024-4 Modern Oxidation Methods 2004 de Vries, J. G., Elsevier, C. J. (eds.) ISBN: 978-3-527-30642-8 The Handbook of Homogeneous Hydrogenation 2007 ISBN: 978-3-527-31161-3 Modern Reduction Methods Edited by Pher G. Andersson and Ian J. Munslow The Editors All books published by Wiley-VCH are carefully produced. Nevertheless, authors, editors, and Prof. Dr. Pher G. Andersson publisher do not warrant the information Uppsala University contained in these books, including this book, to Department of Organic Chemistry be free of errors. Readers are advised to keep in Husargatan 3 mind that statements, data, illustrations, 751 23 Uppsala procedural details or other items may Sweden inadvertently be inaccurate. Dr. Ian J. Munslow Library of Congress Card No.: Uppsala University applied for Department of Biochemistry and Organic Chemistry Husargatan 3 British Library Cataloguing-in-Publication Data 751 23 Uppsala A catalogue record for this book is available from Sweden the British Library. Bibliographic information published by the Deutsche Nationalbibliothek Die Deutsche Nationalbibliothek lists this publication in the Deutsche Nationalbibliografi e; detailed bibliographic data are available on the Internet at <http://dnb.d-nb.de>. -

Phospholipid:Diacylglycerol Acyltransferase: an Enzyme That Catalyzes the Acyl-Coa-Independent Formation of Triacylglycerol in Yeast and Plants
Phospholipid:diacylglycerol acyltransferase: An enzyme that catalyzes the acyl-CoA-independent formation of triacylglycerol in yeast and plants Anders Dahlqvist*†‡, Ulf Ståhl†§, Marit Lenman*, Antoni Banas*, Michael Lee*, Line Sandager¶, Hans Ronne§, and Sten Stymne¶ *Scandinavian Biotechnology Research (ScanBi) AB, Herman Ehles Va¨g 2 S-26831 Svaloˆv, Sweden; ¶Department of Plant Breeding Research, Swedish University of Agricultural Sciences, Herman Ehles va¨g 2–4, S-268 31 Svalo¨v, Sweden; and §Department of Plant Biology, Uppsala Genetic Center, Swedish University of Agricultural Sciences, Box 7080, S-750 07 Uppsala, Sweden Edited by Christopher R. Somerville, Carnegie Institution of Washington, Stanford, CA, and approved March 31, 2000 (received for review February 15, 2000) Triacylglycerol (TAG) is known to be synthesized in a reaction that acid) and epoxidated fatty acid (vernolic acid) in TAG in castor uses acyl-CoA as acyl donor and diacylglycerol (DAG) as acceptor, bean (Ricinus communis) and the hawk’s-beard Crepis palaestina, and which is catalyzed by the enzyme acyl-CoA:diacylglycerol respectively. Furthermore, a similar enzyme is shown to be acyltransferase. We have found that some plants and yeast also present in the yeast Saccharomyces cerevisiae, and the gene have an acyl-CoA-independent mechanism for TAG synthesis, encoding this enzyme, YNR008w, is identified. which uses phospholipids as acyl donors and DAG as acceptor. This reaction is catalyzed by an enzyme that we call phospholipid:dia- Materials and Methods cylglycerol acyltransferase, or PDAT. PDAT was characterized in Yeast Strains and Plasmids. The wild-type yeast strains used were microsomal preparations from three different oil seeds: sunflower, either FY1679 (MAT␣ his3-⌬200 leu2-⌬1 trp1-⌬6 ura3-52) (9) or castor bean, and Crepis palaestina. -

Lanthanide Replacement in Organic Synthesis: Calcium-Mediated Luche-Type Reduction of Α,Β-Functionalised Ketones
Lanthanide Replacement in Organic Synthesis: Calcium-mediated Luche-type Reduction of α,β-functionalised Ketones A thesis submitted by Nina Viktoria Forkel In partial fulfilment of the requirement for a degree of Doctor of Philosophy Imperial College London Department of Chemistry South Kensington Campus SW7 2AZ London United Kingdom September 2013 Lanthanide Replacement in Organic Synthesis: Calcium-mediated Luche-type Reduction of α,β-functionalised Ketones “Man kann sich die Weite und Möglichkeiten des Lebens gar nicht unerschöpflich genug denken.” (Rainer Maria Rilke) This thesis is dedicated to all my loved ones. Declaration of originality and statement of copyright Declaration of originality I, Nina V. Forkel, certify that the research described within this thesis was carried out in the Department of Chemistry at Imperial College London between October 2009 and September 2012. It was accomplished under the primary supervision of Dr Matthew J. Fuchter, Imperial College London, along with supervision from Dr David A. Henderson, Pfizer Ltd. The entire body of work is that of the author, unless otherwise stated to the contrary, and has not been submitted previously for a degree at this or any other university. Statement of copyright The copyright of this thesis rests with the author and is made available under a Creative Commons Attribution Non-Commercial No Derivatives licence. Researchers are free to copy, distribute or transmit the thesis on the condition that they attribute it, that they do not use it for commercial purposes, and that they do not alter, transform or build upon it. For any reuse or redistribution, researchers must make clear to others the licence terms of this work. -

United States Patent (19) 11 Patent Number: 6,057,352 Brown Et Al
US006057352A United States Patent (19) 11 Patent Number: 6,057,352 Brown et al. (45) Date of Patent: May 2, 2000 54 FUNGICIDAL CYCLIC AMIDES WO 96/17851 6/1996 WIPO ............................... CO7F 7/08 WO 96/26.191 8/1996 WIPO ... ... CO7D 249/12 75 Inventors: Richard James Brown, Newark, Del.: WO 96/36633 11/1996 WIPO ... ... CO7D 405/04 Deborah Ann Frasier, Martinez, Calif.; WO96/36229 11/1996 WIPO ... ... A01N 43/653 Michael Henry Howard, Jr., WO 97/02255 1/1997 WIPO m CO7D 261/12 Rockland; Gerard Michael Koether, OTHER PUBLICATIONS Bear, both of Del. Zvilichovsky, G., J. Heterocyclic Chem., 24,465–470, 1987. 73 Assignee: E. I. du Pont de Nemours and Zvilichovsky, G. et al., J. Heterocyclic Chem., 25, Company, Wilmington, Del. 1307-1310, 1988. Davis, M. et al., Australian J. Chem., 30(8), 1815-1818, 21 Appl. No.: 08/952,380 1977. 22 PCT Filed: May 8, 1996 Primary Examiner Mukund J. Shah 86 PCT No.: PCT/US96/06534 Assistant Examiner Deepak R. Rao S371 Date: Nov. 13, 1997 57 ABSTRACT S 102(e) Date: Nov. 13, 1997 Compounds of Formula (I), 87 PCT Pub. No.: WO96/36616 (I) PCT Pub. Date: Nov. 21, 1996 1.Y. Nz Related U.S. Application Data x - W 63 Continuation-in-part of application No. 08/442,433, May NY 17, 1995, abandoned. A-N 60 Provisional application No. 60/004,183, Sep. 22, 1995. Ye 51) Int. Cl." ...................... C07D 249/12; CO7D 233/30; AO1N 43/74; AO1N 43/56 and their N-oxides and agriculturally Suitable Salts are 52 U.S. -

2 Reactions Observed with Alkanes Do Not Occur with Aromatic Compounds 2 (SN2 Reactions Never Occur on Sp Hybridized Carbons!)
Reactions of Aromatic Compounds Aromatic compounds are stabilized by this “aromatic stabilization” energy Due to this stabilization, normal SN2 reactions observed with alkanes do not occur with aromatic compounds 2 (SN2 reactions never occur on sp hybridized carbons!) In addition, the double bonds of the aromatic group do not behave similar to alkene reactions Aromatic Substitution While aromatic compounds do not react through addition reactions seen earlier Br Br Br2 Br2 FeBr3 Br With an appropriate catalyst, benzene will react with bromine The product is a substitution, not an addition (the bromine has substituted for a hydrogen) The product is still aromatic Electrophilic Aromatic Substitution Aromatic compounds react through a unique substitution type reaction Initially an electrophile reacts with the aromatic compound to generate an arenium ion (also called sigma complex) The arenium ion has lost aromatic stabilization (one of the carbons of the ring no longer has a conjugated p orbital) Electrophilic Aromatic Substitution In a second step, the arenium ion loses a proton to regenerate the aromatic stabilization The product is thus a substitution (the electrophile has substituted for a hydrogen) and is called an Electrophilic Aromatic Substitution Energy Profile Transition states Transition states Intermediate Potential E energy H Starting material Products E Reaction Coordinate The rate-limiting step is therefore the formation of the arenium ion The properties of this arenium ion therefore control electrophilic aromatic substitutions (just like any reaction consider the stability of the intermediate formed in the rate limiting step) 1) The rate will be faster for anything that stabilizes the arenium ion 2) The regiochemistry will be controlled by the stability of the arenium ion The properties of the arenium ion will predict the outcome of electrophilic aromatic substitution chemistry Bromination To brominate an aromatic ring need to generate an electrophilic source of bromine In practice typically add a Lewis acid (e.g. -

Organic Chemistry/Fourth Edition: E-Text
CHAPTER 17 ALDEHYDES AND KETONES: NUCLEOPHILIC ADDITION TO THE CARBONYL GROUP O X ldehydes and ketones contain an acyl group RC± bonded either to hydrogen or Ato another carbon. O O O X X X HCH RCH RCRЈ Formaldehyde Aldehyde Ketone Although the present chapter includes the usual collection of topics designed to acquaint us with a particular class of compounds, its central theme is a fundamental reaction type, nucleophilic addition to carbonyl groups. The principles of nucleophilic addition to alde- hydes and ketones developed here will be seen to have broad applicability in later chap- ters when transformations of various derivatives of carboxylic acids are discussed. 17.1 NOMENCLATURE O X The longest continuous chain that contains the ±CH group provides the base name for aldehydes. The -e ending of the corresponding alkane name is replaced by -al, and sub- stituents are specified in the usual way. It is not necessary to specify the location of O X the ±CH group in the name, since the chain must be numbered by starting with this group as C-1. The suffix -dial is added to the appropriate alkane name when the com- pound contains two aldehyde functions.* * The -e ending of an alkane name is dropped before a suffix beginning with a vowel (-al) and retained be- fore one beginning with a consonant (-dial). 654 Back Forward Main Menu TOC Study Guide TOC Student OLC MHHE Website 17.1 Nomenclature 655 CH3 O O O O CH3CCH2CH2CH CH2 CHCH2CH2CH2CH HCCHCH CH3 4,4-Dimethylpentanal 5-Hexenal 2-Phenylpropanedial When a formyl group (±CHœO) is attached to a ring, the ring name is followed by the suffix -carbaldehyde. -

Metabolic Carbonyl Reduction of Anthracyclines — Role in Cardiotoxicity and Cancer Resistance
Invest New Drugs DOI 10.1007/s10637-017-0443-2 REVIEW Metabolic carbonyl reduction of anthracyclines — role in cardiotoxicity and cancer resistance. Reducing enzymes as putative targets for novel cardioprotective and chemosensitizing agents Kamil Piska1 & Paulina Koczurkiewicz1 & Adam Bucki 2 & Katarzyna Wójcik-Pszczoła1 & Marcin Kołaczkowski2 & Elżbieta Pękala1 Received: 23 November 2016 /Accepted: 17 February 2017 # The Author(s) 2017. This article is published with open access at Springerlink.com Summary Anthracycline antibiotics (ANT), such as doxoru- monoHER, curcumin, (−)-epigallocatechin gallate, resvera- bicin or daunorubicin, are a class of anticancer drugs that are trol, berberine or pixantrone, and their modulating effect on widely used in oncology. Although highly effective in cancer the activity of ANT is characterized and discussed as potential therapy, their usefulness is greatly limited by their mechanism of action for novel therapeutics in cancer cardiotoxicity. Possible mechanisms of ANT cardiotoxicity treatment. include their conversion to secondary alcohol metabolites (i.e. doxorubicinol, daunorubicinol) catalyzed by carbonyl re- Keywords Anthracyclines . Cardiotoxicity . Resistance . ductases (CBR) and aldo-keto reductases (AKR). These me- Pharmacokinetics . Drug metabolism . Anticancer agents tabolites are suspected to be more cardiotoxic than their parent compounds. Moreover, overexpression of ANT-reducing en- zymes (CBR and AKR) are found in many ANT-resistant Introduction cancers. The secondary metabolites show decreased cytotoxic properties and are more susceptible to ABC-mediated efflux Anthracyclines (ANT) are a class of cell-cycle non-specific than their parent compounds; thus, metabolite formation is anticancer antibiotics that were first isolated from the considered one of the mechanisms of cancer resistance. Streptomyces genus in the early 1960s. -

Reactions of Aromatic Compounds Just Like an Alkene, Benzene Has Clouds of Electrons Above and Below Its Sigma Bond Framework
Reactions of Aromatic Compounds Just like an alkene, benzene has clouds of electrons above and below its sigma bond framework. Although the electrons are in a stable aromatic system, they are still available for reaction with strong electrophiles. This generates a carbocation which is resonance stabilized (but not aromatic). This cation is called a sigma complex because the electrophile is joined to the benzene ring through a new sigma bond. The sigma complex (also called an arenium ion) is not aromatic since it contains an sp3 carbon (which disrupts the required loop of p orbitals). Ch17 Reactions of Aromatic Compounds (landscape).docx Page1 The loss of aromaticity required to form the sigma complex explains the highly endothermic nature of the first step. (That is why we require strong electrophiles for reaction). The sigma complex wishes to regain its aromaticity, and it may do so by either a reversal of the first step (i.e. regenerate the starting material) or by loss of the proton on the sp3 carbon (leading to a substitution product). When a reaction proceeds this way, it is electrophilic aromatic substitution. There are a wide variety of electrophiles that can be introduced into a benzene ring in this way, and so electrophilic aromatic substitution is a very important method for the synthesis of substituted aromatic compounds. Ch17 Reactions of Aromatic Compounds (landscape).docx Page2 Bromination of Benzene Bromination follows the same general mechanism for the electrophilic aromatic substitution (EAS). Bromine itself is not electrophilic enough to react with benzene. But the addition of a strong Lewis acid (electron pair acceptor), such as FeBr3, catalyses the reaction, and leads to the substitution product. -

Using Dynamic Combinatorial Chemistry to Construct Novel Thioester and Disulfide Lipids
Using dynamic combinatorial chemistry to construct novel thioester and disulfide lipids A dissertation submitted to the University of Manchester for the degree of MSc by Research Chemistry In the Faculty of Science and Engineering 2017 Jack A. Yung School of Chemistry Table of Contents List of figures, tables and equations 5 Symbols and abbreviations 10 Abstract 12 Declaration 13 Copyright statement 13 Acknowledgements 14 The author 14 Chapter 1. Introduction 15 1.1 The cell membrane 16 1.1.1 Function and composition 16 1.2 Membrane lipids 16 1.2.1 Lipid classification 16 1.2.2 Amphiphiles 17 1.2.3 Glycolipids 17 1.2.4 Sterols 18 1.2.5 Phospholipids 19 1.3 Lipid vesicles 20 1.3.1 Supramolecular self-assembly 20 1.3.2 Interaction free energies 21 1.3.3 Framework for the theory of self-assembly 22 1.3.4 Micelles 23 1.3.5 Lipid bilayers 24 1.3.6 Vesicles 25 1.3.7 Phase-transition temperature 27 1.4 Amphiphilic building blocks 27 1.5 Thioesters 29 1.5.1 Thioester reactivity 29 1.5.2 Trans-thioesterification 30 1.5.3 Thioester exchange reactions in DCC 31 1.6 Disulfides 32 2 1.6.1 Disulfide reactivity 32 1.6.2 Thiol-disulfide interchange reactions 32 1.6.3 Disulfide exchange reactions in DCC 33 1.7 Pre-biotic lipids 34 1.7.1 Sources of pre-biotic organic compounds 34 1.7.2 The first pre-biotic membrane structure 36 1.7.3 The role of sulfur in pre-biotic chemistry 36 1.8 Artificially designed vesicles 37 1.8.1 Applications of artificially designed vesicles 37 1.8.2 Zeta-potential 37 1.8.3 Vesicle design 38 1.9 Targets 39 1.9.1 Aims 39 Chapter 2. -
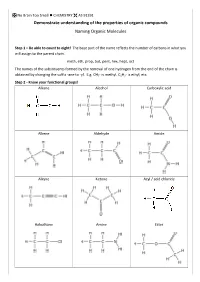
IUPAC Naming
No Brain Too Small CHEMISTRY AS 91391 Demonstrate understanding of the properties of organic compounds Naming Organic Molecules Step 1 – Be able to count to eight! The base part of the name reflects the number of carbons in what you will assign to the parent chain. meth, eth, prop, but, pent, hex, hept, oct The names of the substituents formed by the removal of one hydrogen from the end of the chain is obtained by changing the suffix -ane to -yl. E.g. CH3- is methyl, C2H5- is ethyl, etc. Step 2 - Know your functional groups! Alkane Alcohol Carboxylic acid Alkene Aldehyde Amide Alkyne Ketone Acyl / acid chloride Haloalkane Amine Ester No Brain Too Small CHEMISTRY AS 91391 This is NOT an exhaustive list of rules but a guide for L3 NCEA. Identify the principle functional group in the structure. If there is only ONE functional group then this is the principle functional group e.g. CH3CH2CH2COOH will have the suffix -oic acid as the carboxylic acid is the principle functional group. In cases where compounds have more than one functional group, then the principle functional group is decided by a priority order. carboxylic acids > acid derivatives* > aldehydes > ketones > alcohols > amines *esters, acyl/acid chlorides and amides 3-hydroxybutanoic acid 4-aminopentanoic acid as the carboxylic acid functional group takes priority Fluoro-, chloro-, bromo-, iodo- and alkyl groups have “no priority”. Their numbering is governed by the lowest sum rule. 2,3-dichloro-4-methylhexane and NOT 4,5-dichloro-2-methylhexane (as 2+3+4 = 9 whereas 2+4+5 = 11) No Brain Too Small CHEMISTRY AS 91391 Rules 1. -

Chem 314 Preorganic Evaluation
Organic Reaction Guide Beauchamp 1 Chem 316 / Beauchamp Reactions Review Sheet Name SN2 Reactions - special features: biomolecular kinetics Rate = kSN2[RX][Nu ], single step concerted reaction, E2 is a competing reaction o o o o relative order of reactivity: CH3X > 1 RX > 2 RX >> 3 RX (based on steric hinderance, no SN2 at 3 RX) allylic & benzylic RX are very reactive, adjacent pi bonds help stabilize transition state and lower TS energy (Ea) o complete substitution at Cα (3 RX) or Cβ (neopentyl pattern) almost completely inhibits SN2 reactions vinyl & phenyl are very unreactive, bonds are stronger and poor backside approach leaving group ability: OTs = I > Br > Cl in neutral or basic conditions (just like E2, SN1 adn E1), and neutral molecule leaving groups are good from protonated, cationic intermediates in acid conditions, + + + + -OH2 , -ORH , -OR2 , -NR3 , etc. we will consider all anions, ammonia, amines, thiols and sulfides to be strong nucleophiles (favors SN2 and E2 reactions) in our course some electron pair donors are mainly nucleophiles (sulfur, azide, cyanide, carboxylates) and - + + - + - some are mainly bases (t-BuO K , Na H2N , Na H ) polar, aprotic solvents work best for SN2 reactions because nucleophiles are relatively unencombered for electron doantion (dimethyl sulofoxide = DMSO, dimethylformamide = DMF, acetonitrile = AN, acetone, etc.) in our course some electron pair donors are mainly nucleophiles (sulfur, azide, cyanide, carboxylates) and we will consider neutral solvent molecules such as water, alcohols and acids to be weak nucleophiles (favors SN1 and E1) stereoselectivity: 100% inversion of configuration from backside atack regioselectivity: reacts at carbon with leaving group, completely unambiguous chemoselectivity: N/A The following list is designed to emphasize SN2 reactions.