Production of High-Purity Radium-223 from Legacy Actinium-Beryllium Neutron Sources†
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Development of the Periodic Table and Its Consequences Citation: J
Firenze University Press www.fupress.com/substantia The Development of the Periodic Table and its Consequences Citation: J. Emsley (2019) The Devel- opment of the Periodic Table and its Consequences. Substantia 3(2) Suppl. 5: 15-27. doi: 10.13128/Substantia-297 John Emsley Copyright: © 2019 J. Emsley. This is Alameda Lodge, 23a Alameda Road, Ampthill, MK45 2LA, UK an open access, peer-reviewed article E-mail: [email protected] published by Firenze University Press (http://www.fupress.com/substantia) and distributed under the terms of the Abstract. Chemistry is fortunate among the sciences in having an icon that is instant- Creative Commons Attribution License, ly recognisable around the world: the periodic table. The United Nations has deemed which permits unrestricted use, distri- 2019 to be the International Year of the Periodic Table, in commemoration of the 150th bution, and reproduction in any medi- anniversary of the first paper in which it appeared. That had been written by a Russian um, provided the original author and chemist, Dmitri Mendeleev, and was published in May 1869. Since then, there have source are credited. been many versions of the table, but one format has come to be the most widely used Data Availability Statement: All rel- and is to be seen everywhere. The route to this preferred form of the table makes an evant data are within the paper and its interesting story. Supporting Information files. Keywords. Periodic table, Mendeleev, Newlands, Deming, Seaborg. Competing Interests: The Author(s) declare(s) no conflict of interest. INTRODUCTION There are hundreds of periodic tables but the one that is widely repro- duced has the approval of the International Union of Pure and Applied Chemistry (IUPAC) and is shown in Fig.1. -

FRANCIUM Element Symbol: Fr Atomic Number: 87
FRANCIUM Element Symbol: Fr Atomic Number: 87 An initiative of IYC 2011 brought to you by the RACI KAYE GREEN www.raci.org.au FRANCIUM Element symbol: Fr Atomic number: 87 Francium (previously known as eka-cesium and actinium K) is a radioactive metal and the second rarest naturally occurring element after Astatine. It is the least stable of the first 103 elements. Very little is known of the physical and chemical properties of Francium compared to other elements. Francium was discovered by Marguerite Perey of the Curie Institute in Paris, France in 1939. However, the existence of an element of atomic number 87 was predicted in the 1870s by Dmitri Mendeleev, creator of the first version of the periodic table, who presumed it would have chemical and physical properties similar to Cesium. Several research teams attempted to isolate this missing element, and there were at least four false claims of discovery during which it was named Russium (after the home country of soviet chemist D. K. Dobroserdov), Alkalinium (by English chemists Gerald J. K. Druce and Frederick H. Loring as the heaviest alkali metal), Virginium (after Virginia, home state of chemist Fred Allison), and Moldavium (by Horia Hulubei and Yvette Cauchois after Moldavia, the Romanian province where they conducted their work). Perey finally discovered Francium after purifying radioactive Actinium-227 from Lanthanum, and detecting particles decaying at low energy levels not previously identified. The new product exhibited chemical properties of an alkali metal (such as co-precipitating with Cesium salts), which led Perey to believe that it was element 87, caused by the alpha radioactive decay of Actinium-227. -

The Separation of Bismuth-213 from Actinium-225 and the Ion Exchange Properties of the Alkali Metal Cations with an Inorganic Resin
University of Tennessee, Knoxville TRACE: Tennessee Research and Creative Exchange Doctoral Dissertations Graduate School 12-2017 The Separation of Bismuth-213 from Actinium-225 and the Ion Exchange Properties of the Alkali Metal Cations with an Inorganic Resin Mark Alan Moore University of Tennessee Follow this and additional works at: https://trace.tennessee.edu/utk_graddiss Recommended Citation Moore, Mark Alan, "The Separation of Bismuth-213 from Actinium-225 and the Ion Exchange Properties of the Alkali Metal Cations with an Inorganic Resin. " PhD diss., University of Tennessee, 2017. https://trace.tennessee.edu/utk_graddiss/4848 This Dissertation is brought to you for free and open access by the Graduate School at TRACE: Tennessee Research and Creative Exchange. It has been accepted for inclusion in Doctoral Dissertations by an authorized administrator of TRACE: Tennessee Research and Creative Exchange. For more information, please contact [email protected]. To the Graduate Council: I am submitting herewith a dissertation written by Mark Alan Moore entitled "The Separation of Bismuth-213 from Actinium-225 and the Ion Exchange Properties of the Alkali Metal Cations with an Inorganic Resin." I have examined the final electronic copy of this dissertation for form and content and recommend that it be accepted in partial fulfillment of the equirr ements for the degree of Doctor of Philosophy, with a major in Chemical Engineering. Robert Counce, Major Professor We have read this dissertation and recommend its acceptance: Paul Dalhaimer, Howard Hall, George Schweitzer, Jack Watson Accepted for the Council: Dixie L. Thompson Vice Provost and Dean of the Graduate School (Original signatures are on file with official studentecor r ds.) The Separation of Bismuth-213 from Actinium-225 and the Ion Exchange Properties of the Alkali Metal Cations with an Inorganic Resin A Dissertation Presented for the Doctor of Philosophy Degree The University of Tennessee, Knoxville Mark Alan Moore December 2017 Copyright © 2017 by Mark A. -
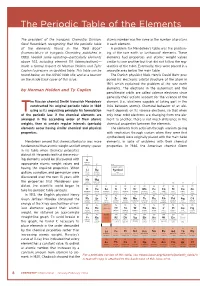
The Periodic Table of the Elements
The Periodic Table of the Elements The president of the Inorganic Chemistry Division, atomic number was the same as the number of protons Gerd Rosenblatt, recognizing that the periodic table in each element. of the elements found in the “Red Book” A problem for Mendeleev’s table was the position- (Nomenclature of Inorganic Chemistry, published in ing of the rare earth or lanthanoid* elements. These 1985) needed some updating—particularly elements elements had properties and atomic weight values above 103, including element 110 (darmstadtium)— similar to one another but that did not follow the reg- made a formal request to Norman Holden and Tyler ularities of the table. Eventually, they were placed in a Coplen to prepare an updated table. This table can be separate area below the main table. found below, on the IUPAC Web site, and as a tear-off The Danish physicist Niels Henrik David Bohr pro- on the inside back cover of this issue. posed his electronic orbital structure of the atom in 1921, which explained the problem of the rare earth by Norman Holden and Ty Coplen elements. The electrons in the outermost and the penultimate orbits are called valence electrons since generally their actions account for the valence of the he Russian chemist Dmitri Ivanovich Mendeleev element (i.e., electrons capable of taking part in the constructed his original periodic table in 1869 links between atoms). Chemical behavior of an ele- Tusing as its organizing principle his formulation ment depends on its valence electrons, so that when of the periodic law: if the chemical elements are only inner orbit electrons are changing from one ele- arranged in the ascending order of their atomic ment to another, there is not much difference in the weights, then at certain regular intervals (periods) chemical properties between the elements. -

Periodic Table of the Elements Notes
Periodic Table of the Elements Notes Arrangement of the known elements based on atomic number and chemical and physical properties. Divided into three basic categories: Metals (left side of the table) Nonmetals (right side of the table) Metalloids (touching the zig zag line) Basic Organization by: Atomic structure Atomic number Chemical and Physical Properties Uses of the Periodic Table Useful in predicting: chemical behavior of the elements trends properties of the elements Atomic Structure Review: Atoms are made of protons, electrons, and neutrons. Elements are atoms of only one type. Elements are identified by the atomic number (# of protons in nucleus). Energy Levels Review: Electrons are arranged in a region around the nucleus called an electron cloud. Energy levels are located within the cloud. At least 1 energy level and as many as 7 energy levels exist in atoms Energy Levels & Valence Electrons Energy levels hold a specific amount of electrons: 1st level = up to 2 2nd level = up to 8 3rd level = up to 8 (first 18 elements only) The electrons in the outermost level are called valence electrons. Determine reactivity - how elements will react with others to form compounds Outermost level does not usually fill completely with electrons Using the Table to Identify Valence Electrons Elements are grouped into vertical columns because they have similar properties. These are called groups or families. Groups are numbered 1-18. Group numbers can help you determine the number of valence electrons: Group 1 has 1 valence electron. Group 2 has 2 valence electrons. Groups 3–12 are transition metals and have 1 or 2 valence electrons. -
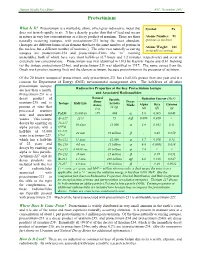
Protactinium
Human Health Fact Sheet ANL, November 2001 Protactinium What Is It? Protactinium is a malleable, shiny, silver-gray radioactive metal that Symbol: Pa does not tarnish rapidly in air. It has a density greater than that of lead and occurs in nature in very low concentrations as a decay product of uranium. There are three Atomic Number: 91 naturally occurring isotopes, with protactinium-231 being the most abundant. (protons in nucleus) (Isotopes are different forms of an element that have the same number of protons in Atomic Weight: 231 the nucleus but a different number of neutrons.) The other two naturally occurring (naturally occurring) isotopes are protactinium-234 and protactinium-234m (the “m” meaning metastable), both of which have very short half-lives (6.7 hours and 1.2 minutes, respectively) and occur in extremely low concentrations. Protactinium was first identified in 1913 by Kasimir Fajans and O.H. Gohring (as the isotope protactinium-234m), and protactinium-231 was identified in 1917. The name comes from the Greek work protos (meaning first) and the element actinium, because protactinium is the precursor of actinium. Of the 20 known isotopes of protactinium, only protactinium-231 has a half-life greater than one year and is a concern for Department of Energy (DOE) environmental management sites. The half-lives of all other protactinium isotopes Radioactive Properties of the Key Protactinium Isotope are less than a month. Protactinium-231 is a and Associated Radionuclides Natural decay product of Specific Radiation Energy (MeV) Abun- Decay uranium-235 and is Isotope Half-Life Activity dance Mode Alpha Beta Gamma present at sites that (Ci/g) (%) (α) (β) (γ) processed uranium α ores and associated Pa231 33,000 yr >99 .048 5.0 0.065 0.048 wastes. -

Genius of the Periodic Table
GENIUS OF THE PERIODIC TABLE "Isn't it the work of a genius'. " exclaimed Academician V.I. Spitsyn, USSR, a member of the Scientific Advisory Committee when talking to an Agency audience in January. His listeners shared his enthusiasm. Academician Spitsyn was referring to the to the first formulation a hundred years ago by Professor Dmitry I. Mendeleyev of the Periodic Law of Elements. In conditions of enormous difficulty, considering the lack of data on atomic weights of elements, Mendeleyev created in less than two years work at St. Petersburg University, a system of chemical elements that is, in general, still being used. His law became a powerful instrument for further development of chemistry and physics. He was able immediately to correct the atomic weight numbers of some elements, including uranium, whose atomic weight he found to be double that given at the time. Two years later Mendeleyev went so far as to give a detailed description of physical or chemical properties of some elements which were as yet undiscovered. Time gave striking proof of his predictions and his periodic law. Mendeleyev published his conclusions in the first place by sending, early in March 186 9, a leaflet to many Russian and foreign scientists. It gave his system of elements based on their atomic weights and chemical resemblance. On the 18th March that year his paper on the subject was read at the meeting of the Russian Chemical Society, and two months later the Society's Journal published his article entitled "The correlation between properties of elements and their atomic weight". -

Periodic Table P J STEWART / SCIENCE PHOTO LIBRARY PHOTO SCIENCE / STEWART J P
Periodic table P J STEWART / SCIENCE PHOTO LIBRARY PHOTO SCIENCE / STEWART J P 46 | Chemistry World | March 2009 www.chemistryworld.org Periodic change The periodic table, cherished by generations of chemists, has steadily evolved over time. Eric Scerri is among those now calling for drastic change The periodic table has become recurrences as vertical columns or something of a style icon while In short groups. remaining indispensable to chemists. In its original form The notion of chemical reactivity Over the years the table has had the periodic table was is something of a vague one. To make to change to accommodate new relatively simple. Over this idea more precise, the periodic elements. But some scientists the years, extra elements table pioneers focused on the propose giving the table a makeover have been added and the maximum valence of each element while others call for drastic changes layout of the transition and looked for similarities among to its core structure. elements altered these quantities (see Mendeleev’s More than 1000 periodic systems Some call for drastic table, p48). have been published since the table rearrangements, The method works very well for Russian chemist Dimitri Mendeleev perhaps placing hydrogen the elements up to atomic weight developed the mature periodic with the halogens. 55 (manganese) after which point system – the most fundamental A new block may be it starts to fall apart. Although natural system of classification needed when chemists there seems to be a repetition in the ever devised. (Not to mention the can make elements in highest valence of aluminium and hundreds if not thousands of new the g-block, starting at scandium (3), silicon and titanium systems that have appeared since the element 121 (4), phosphorus and vanadium (5), advent of the internet.) and chlorine and manganese (7), Such a proliferation prompts this is not the case with potassium questions as to whether some tables and iron. -

The Environmental Behaviour of Polonium
technical reportS series no. 484 Technical Reports SeriEs No. 484 The Environmental Behaviour of Polonium F. Carvalho, S. Fernandes, S. Fesenko, E. Holm, B. Howard, The Environmental Behaviour of Polonium P. Martin, M. Phaneuf, D. Porcelli, G. Pröhl, J. Twining @ THE ENVIRONMENTAL BEHAVIOUR OF POLONIUM The following States are Members of the International Atomic Energy Agency: AFGHANISTAN GEORGIA OMAN ALBANIA GERMANY PAKISTAN ALGERIA GHANA PALAU ANGOLA GREECE PANAMA ANTIGUA AND BARBUDA GUATEMALA PAPUA NEW GUINEA ARGENTINA GUYANA PARAGUAY ARMENIA HAITI PERU AUSTRALIA HOLY SEE PHILIPPINES AUSTRIA HONDURAS POLAND AZERBAIJAN HUNGARY PORTUGAL BAHAMAS ICELAND QATAR BAHRAIN INDIA REPUBLIC OF MOLDOVA BANGLADESH INDONESIA ROMANIA BARBADOS IRAN, ISLAMIC REPUBLIC OF RUSSIAN FEDERATION BELARUS IRAQ RWANDA BELGIUM IRELAND SAN MARINO BELIZE ISRAEL SAUDI ARABIA BENIN ITALY SENEGAL BOLIVIA, PLURINATIONAL JAMAICA SERBIA STATE OF JAPAN SEYCHELLES BOSNIA AND HERZEGOVINA JORDAN SIERRA LEONE BOTSWANA KAZAKHSTAN SINGAPORE BRAZIL KENYA SLOVAKIA BRUNEI DARUSSALAM KOREA, REPUBLIC OF SLOVENIA BULGARIA KUWAIT SOUTH AFRICA BURKINA FASO KYRGYZSTAN SPAIN BURUNDI LAO PEOPLE’S DEMOCRATIC SRI LANKA CAMBODIA REPUBLIC SUDAN CAMEROON LATVIA SWAZILAND CANADA LEBANON SWEDEN CENTRAL AFRICAN LESOTHO SWITZERLAND REPUBLIC LIBERIA SYRIAN ARAB REPUBLIC CHAD LIBYA TAJIKISTAN CHILE LIECHTENSTEIN THAILAND CHINA LITHUANIA THE FORMER YUGOSLAV COLOMBIA LUXEMBOURG REPUBLIC OF MACEDONIA CONGO MADAGASCAR TOGO COSTA RICA MALAWI TRINIDAD AND TOBAGO CÔTE D’IVOIRE MALAYSIA TUNISIA CROATIA MALI -

The Elements.Pdf
A Periodic Table of the Elements at Los Alamos National Laboratory Los Alamos National Laboratory's Chemistry Division Presents Periodic Table of the Elements A Resource for Elementary, Middle School, and High School Students Click an element for more information: Group** Period 1 18 IA VIIIA 1A 8A 1 2 13 14 15 16 17 2 1 H IIA IIIA IVA VA VIAVIIA He 1.008 2A 3A 4A 5A 6A 7A 4.003 3 4 5 6 7 8 9 10 2 Li Be B C N O F Ne 6.941 9.012 10.81 12.01 14.01 16.00 19.00 20.18 11 12 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 3 Na Mg IIIB IVB VB VIB VIIB ------- VIII IB IIB Al Si P S Cl Ar 22.99 24.31 3B 4B 5B 6B 7B ------- 1B 2B 26.98 28.09 30.97 32.07 35.45 39.95 ------- 8 ------- 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 4 K Ca Sc Ti V Cr Mn Fe Co Ni Cu Zn Ga Ge As Se Br Kr 39.10 40.08 44.96 47.88 50.94 52.00 54.94 55.85 58.47 58.69 63.55 65.39 69.72 72.59 74.92 78.96 79.90 83.80 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 5 Rb Sr Y Zr NbMo Tc Ru Rh PdAgCd In Sn Sb Te I Xe 85.47 87.62 88.91 91.22 92.91 95.94 (98) 101.1 102.9 106.4 107.9 112.4 114.8 118.7 121.8 127.6 126.9 131.3 55 56 57 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 6 Cs Ba La* Hf Ta W Re Os Ir Pt AuHg Tl Pb Bi Po At Rn 132.9 137.3 138.9 178.5 180.9 183.9 186.2 190.2 190.2 195.1 197.0 200.5 204.4 207.2 209.0 (210) (210) (222) 87 88 89 104 105 106 107 108 109 110 111 112 114 116 118 7 Fr Ra Ac~RfDb Sg Bh Hs Mt --- --- --- --- --- --- (223) (226) (227) (257) (260) (263) (262) (265) (266) () () () () () () http://pearl1.lanl.gov/periodic/ (1 of 3) [5/17/2001 4:06:20 PM] A Periodic Table of the Elements at Los Alamos National Laboratory 58 59 60 61 62 63 64 65 66 67 68 69 70 71 Lanthanide Series* Ce Pr NdPmSm Eu Gd TbDyHo Er TmYbLu 140.1 140.9 144.2 (147) 150.4 152.0 157.3 158.9 162.5 164.9 167.3 168.9 173.0 175.0 90 91 92 93 94 95 96 97 98 99 100 101 102 103 Actinide Series~ Th Pa U Np Pu AmCmBk Cf Es FmMdNo Lr 232.0 (231) (238) (237) (242) (243) (247) (247) (249) (254) (253) (256) (254) (257) ** Groups are noted by 3 notation conventions. -

Simultaneous Separation of Actinium and Radium Isotopes from a Proton
www.nature.com/scientificreports OPEN Simultaneous Separation of Actinium and Radium Isotopes from a Proton Irradiated Thorium Received: 23 May 2017 Accepted: 19 July 2017 Matrix Published: xx xx xxxx Tara Mastren1, Valery Radchenko1,3, Allison Owens 2, Roy Copping2, Rose Boll 2, Justin R. Griswold2, Saed Mirzadeh2, Lance E. Wyant2, Mark Brugh1, Jonathan W. Engle1,4, Francois M. Nortier1, Eva R. Birnbaum1, Kevin D. John1 & Michael E. Fassbender1 A new method has been developed for the isolation of 223,224,225Ra, in high yield and purity, from a proton irradiated 232Th matrix. Herein we report an all-aqueous process using multiple solid-supported adsorption steps including a citrate chelation method developed to remove >99.9% of the barium contaminants by activity from the fnal radium product. A procedure involving the use of three columns in succession was developed, and the separation of 223,224,225Ra from the thorium matrix was obtained with an overall recovery yield of 91 ± 3%, average radiochemical purity of 99.9%, and production yields that correspond to physical yields based on previously measured excitation functions. Radium-223 chloride (t1/2 11.4d) is available as a U.S. FDA approved pharmaceutical for the treatment of bone metastases under the trademark Xofgo©1. Ionic radium is a bone seeking agent, due to its chemical similarity to calcium. It preferentially accumulates in the rapidly forming cells in bone metastases1, 2. Radium-223 is the frst alpha emitting isotope that obtained FDA approval for the treatment of cancer. Other radium isotopes of interest 224 225 for preclinical research are Ra (t1/2 3.6d) and Ra (t1/2 14.9d). -
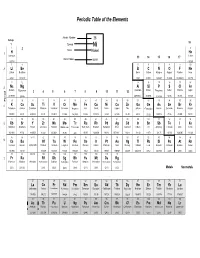
Periodic Table and Atomic Weights
Periodic Table of the Elements Atomic Number 28 Group 18 1 Symbol Ni 1 2 2 Name Nickel 1 H He Hydrogen Helium 13 14 15 16 17 Atomic Mass 58.69 1.00794 4.00260 3 4 5 6 7 8 9 10 2 Li Be B C N O F Ne Lithium Beryllium Boron Carbon Nitrogen Oxygen Fluorine Neon 6.941 9.01218 10.81 12.0111 14.0067 15.9994 18.998403 20.179 11 12 13 14 15 16 17 18 3 Na Mg Al Si P S Cl Ar Sodium Magnesium Aluminum Silicon Phosphorus Sulfur Chlorine Argon 3 4 5 6 7 8 9 10 11 12 22.98977 24.305 26.98154 28.0855 30.97376 32.06 35.453 39.948 Period 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 4 K Ca Sc Ti V Cr Mn Fe Co Ni Cu Zn Ga Ge As Se Br Kr Potassium Calcium Scandium Titanium Vanadium Chromium Manganese Iron Cobalt Nickel Copper Zinc Gallium Germanium Arsenic Selenium Bromine Krypton 39.0983 40.08 44.9559 47.88 50.9415 51.996 54.9380 55.847 58.9332 58.69 63.546 65.39 69.72 72.59 74.9216 78.96 79.904 83.80 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 5 Rb Sr Y Zr Nb Mo Tc Ru Rh Pd Ag Cd In Sn Sb Te I Xe Rubidium Strontium Yttrium Zirconium Niobium Molybdenum Technetium Ruthenium Rhodium Palladium Silver Cadmium Indium Tin Antimony Tellurium Iodine Xenon 85.4678 87.62 88.9059 91.224 92.9064 95.94 (98) 101.07 102.906 106.42 107.868 112.41 114.82 118.71 121.75 127.60 126.905 131.29 55 56 57-71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 6 Cs Ba Hf Ta W Re Os Ir Pt Au Hg Tl Pb Bi Po At Rn Caesium Barium Lanthanoids Hafnium Tantalum Tungsten Rhenium Osmium Iridium Platinum Gold Mercury Thallium Lead Bismuth Polonium Astatine Radon 132.905 137.33