Comparing DNA Yield from Sh Scales Following Different Extraction Protocols
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

TNP SOK 2011 Internet
GARDEN ROUTE NATIONAL PARK : THE TSITSIKAMMA SANP ARKS SECTION STATE OF KNOWLEDGE Contributors: N. Hanekom 1, R.M. Randall 1, D. Bower, A. Riley 2 and N. Kruger 1 1 SANParks Scientific Services, Garden Route (Rondevlei Office), PO Box 176, Sedgefield, 6573 2 Knysna National Lakes Area, P.O. Box 314, Knysna, 6570 Most recent update: 10 May 2012 Disclaimer This report has been produced by SANParks to summarise information available on a specific conservation area. Production of the report, in either hard copy or electronic format, does not signify that: the referenced information necessarily reflect the views and policies of SANParks; the referenced information is either correct or accurate; SANParks retains copies of the referenced documents; SANParks will provide second parties with copies of the referenced documents. This standpoint has the premise that (i) reproduction of copywrited material is illegal, (ii) copying of unpublished reports and data produced by an external scientist without the author’s permission is unethical, and (iii) dissemination of unreviewed data or draft documentation is potentially misleading and hence illogical. This report should be cited as: Hanekom N., Randall R.M., Bower, D., Riley, A. & Kruger, N. 2012. Garden Route National Park: The Tsitsikamma Section – State of Knowledge. South African National Parks. TABLE OF CONTENTS 1. INTRODUCTION ...............................................................................................................2 2. ACCOUNT OF AREA........................................................................................................2 -

Professor Catriona Macleod
RRR | Cover 2015 v2 11/9/16 10:17 AM Page 1 C M Y CM MY CY CMY K Composite RRR 2015 | Features 11/12/16 1:36 PM Page 1 C M Y CM MY CY CMY K RHODES UNIVERSITY RESEARCH REPORT A publication of the Rhodes University Research Office, compiled and edited by Tarryn Gillitt, Busi Goba, Patricia Jacob, Jill Macgregor and Jaine Roberts Design & Layout: Sally Dore Research Office Director: Jaine Roberts [email protected] Tel: +27 (46) 603 8756/7572 www.ru.ac.za Cover: Rhodes University researchers Pam Maseko, Nomalanga Mkhize, Heila Lotz-Sisitka, Ruth Simbao, Anthea Garman and Catriona Macleod Cover Photos: Paul Greenway/www.3pphotography.com RESEARCH REPORT 2015 Composite RRR 2015 | Features 11/12/16 1:36 PM Page 2 C M Y CM MY CY CMY K CONTENTS 01 FOREWORD Dr Sizwe Mabizela, Vice-Chancellor 03 INTRODUCTION Dr Peter Clayton, Deputy Vice-Chancellor: Research & Development 05 TOP 30 RESEARCHERS 06 PHD GRADUATES 11 VICE-CHANCELLOR’S BOOK AWARD Professor Anthea Garman 13 VICE-CHANCELLOR’S DISTINGUISHED SENIOR RESEARCH AWARD Professor Catriona Macleod 15 VICE-CHANCELLOR’S DISTINGUISHED RESEARCH AWARD Dr Adrienne Edkins 17 SARChI CHAIRS Professor Heila Lotz-Sisitka, Professor Ruth Simbao and Dr Adrienne Edkins 23 AFRICAN LANGUAGES, SCHOOL OF LANGUAGES AND LITERATURE Associate Professor Pamela Maseko 25 DEPARTMENT OF HISTORY Dr Nomalanga Mkhize RESEARCH REPORT 2015 Composite RRR 2015 | Features 11/12/16 1:34 PM Page 3 C M Y CM MY CY CMY K RHODES RESEARCH 2015 RESEARCH REPORT DEPARTMENT PUBLICATIONS AFFILIATES, INSTITUTES AND 28 Publications from the Vice-Chancellorate -

Western Indian Ocean Marine Science Association Scientific Symposium
WESTERN th INDIAN OCEAN MARINE SCIENCE ASSOCIATION SCIENTIFIC 9 SYMPOSIUM 26th October – 31st October 2015 Wild Coast Sun Resort Eastern Cape, South Africa WESTERN INDIAN OCEAN MARINE SCIENCE ASSOCIATION SCIENTIFIC SYMPOSIUM 26th October – 31st October 2015 Wild Coast Sun Resort Eastern Cape, South Africa SPONSORS:: Coastal, Marine and Island Specific Biodiversity Management in ESA-I0 Coastal States Cover photo credits: Oceanographic Research Instittute (ORI) Design by: G. Arara 9th WIOMSA Scientific Symposium ii TABLE OF CONTENTS SPONSORS ....................................................................................................................................ii OUTLINE OF THE SYMPOSIUM PROGRAMME ..................................................................... 2 MEMBERS OF THE SYMPOSIUM SCIENTIFIC COMMITTEE .............................................. 3 INTRODUCTION .......................................................................................................................... 4 SYMPOSIUM ROOM PLAN ........................................................................................................ 7 SESSION CHAIRS AND RAPPOURTEURS ............................................................................... 8 SCIENTIFIC PROGRAMME ........................................................................................................ 9 POSTERS ..................................................................................................................................... 19 PRE-SYMPOSIUM EVENTS ..................................................................................................... -

First Record of the Twobar Sea Bream Acanthopagrus Bifasciatus (Teleostei: Sparidae) in the Mediterranean Sea
Mediterranean Marine Science Vol. 15, 2014 First record of the twobar sea bream Acanthopagrus bifasciatus (Teleostei: Sparidae) in the Mediterranean Sea BEN-SOUISSI J. Institut National Agronomique de Tunisie, 43 avenue Charles Nicolle, cité Mahrajène, 1082 Tunis, Tunisia BOUGHEDIR W. Institut National Agronomique de Tunisie, 43 avenue Charles Nicolle, cité Mahrajène, 1082 Tunis, Tunisia RIFI M. Institut National Agronomique de Tunisie, 43 avenue Charles Nicolle, cité Mahrajène, 1082 Tunis, Tunisia CAPAPE C. Université Montpellier II, Sciences et Techniques du Languedoc, 34 095 Montpellier cedex 5, France AZZURRO E. ISPRA National Institute for Environmental Protection and research https://doi.org/10.12681/mms.774 Copyright © 2014 To cite this article: BEN-SOUISSI, J., BOUGHEDIR, W., RIFI, M., CAPAPE, C., & AZZURRO, E. (2014). First record of the twobar sea bream Acanthopagrus bifasciatus (Teleostei: Sparidae) in the Mediterranean Sea. Mediterranean Marine Science, 15(2), 437-439. doi:https://doi.org/10.12681/mms.774 http://epublishing.ekt.gr | e-Publisher: EKT | Downloaded at 23/05/2020 04:58:03 | Short Communication Mediterranean Marine Science Indexed in WoS (Web of Science, ISI Thomson) and SCOPUS The journal is available on line at http://www.medit-mar-sc.net DOI: http://dx.doi.org/10.12681/mms.774 First record of the twobar sea bream Acanthopagrus bifasciatus (Teleostei: Sparidae) in the Mediterranean Sea J. BEN SOUISSI1, M. RIFI1, R.GHANEM, W. BOUGHEDIR1, C. CAPAPÉ2 and E. AZZURRO3 1 Département des Ressources Animales, Halieutiques -

A Molecular Phylogeny of the Sparidae (Perciformes: Percoidei)
W&M ScholarWorks Dissertations, Theses, and Masters Projects Theses, Dissertations, & Master Projects 2000 A molecular phylogeny of the Sparidae (Perciformes: Percoidei) Thomas M. Orrell College of William and Mary - Virginia Institute of Marine Science Follow this and additional works at: https://scholarworks.wm.edu/etd Part of the Genetics Commons, and the Zoology Commons Recommended Citation Orrell, Thomas M., "A molecular phylogeny of the Sparidae (Perciformes: Percoidei)" (2000). Dissertations, Theses, and Masters Projects. Paper 1539616799. https://dx.doi.org/doi:10.25773/v5-x8gj-1114 This Dissertation is brought to you for free and open access by the Theses, Dissertations, & Master Projects at W&M ScholarWorks. It has been accepted for inclusion in Dissertations, Theses, and Masters Projects by an authorized administrator of W&M ScholarWorks. For more information, please contact [email protected]. INFORMATION TO USERS This manuscript has been reproduced from the microfilm master. UMI films the text directly from (he original or copy submitted. Thus, some thesis and dissertation copies are in typewriter face, while others may be from any type of computer printer. The quality of this reproduction is dependent upon the quality of the copy submitted. Broken or indistinct print, colored or poor quality illustrations and photographs, print bieedthrough, substandard margins, and improper alignment can adversely affect reproduction. In the unlikely event that the author did not send UMI a complete manuscript and there are missing pages, these will be noted. Also, if unauthorized copyright material had to be removed, a note will indicate the deletion. Oversize materials (e.g., maps, drawings, charts) are reproduced by sectioning the original, beginning at the upper left-hand comer and continuing from left to right in equal sections with small overlaps. -
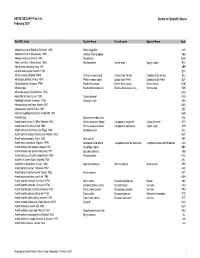
ASFIS ISSCAAP Fish List February 2007 Sorted on Scientific Name
ASFIS ISSCAAP Fish List Sorted on Scientific Name February 2007 Scientific name English Name French name Spanish Name Code Abalistes stellaris (Bloch & Schneider 1801) Starry triggerfish AJS Abbottina rivularis (Basilewsky 1855) Chinese false gudgeon ABB Ablabys binotatus (Peters 1855) Redskinfish ABW Ablennes hians (Valenciennes 1846) Flat needlefish Orphie plate Agujón sable BAF Aborichthys elongatus Hora 1921 ABE Abralia andamanika Goodrich 1898 BLK Abralia veranyi (Rüppell 1844) Verany's enope squid Encornet de Verany Enoploluria de Verany BLJ Abraliopsis pfefferi (Verany 1837) Pfeffer's enope squid Encornet de Pfeffer Enoploluria de Pfeffer BJF Abramis brama (Linnaeus 1758) Freshwater bream Brème d'eau douce Brema común FBM Abramis spp Freshwater breams nei Brèmes d'eau douce nca Bremas nep FBR Abramites eques (Steindachner 1878) ABQ Abudefduf luridus (Cuvier 1830) Canary damsel AUU Abudefduf saxatilis (Linnaeus 1758) Sergeant-major ABU Abyssobrotula galatheae Nielsen 1977 OAG Abyssocottus elochini Taliev 1955 AEZ Abythites lepidogenys (Smith & Radcliffe 1913) AHD Acanella spp Branched bamboo coral KQL Acanthacaris caeca (A. Milne Edwards 1881) Atlantic deep-sea lobster Langoustine arganelle Cigala de fondo NTK Acanthacaris tenuimana Bate 1888 Prickly deep-sea lobster Langoustine spinuleuse Cigala raspa NHI Acanthalburnus microlepis (De Filippi 1861) Blackbrow bleak AHL Acanthaphritis barbata (Okamura & Kishida 1963) NHT Acantharchus pomotis (Baird 1855) Mud sunfish AKP Acanthaxius caespitosa (Squires 1979) Deepwater mud lobster Langouste -

Biology and Fisheries Assessment of the Arabian Pandora (Pagellus Affinis)
BIOLOGY AND FISHERIES ASSESSMENT OF THE ARABIAN PANDORA (Pagellus affinis) (Boulenger, 1887) IN THE ARABIAN SEA, SULTANATE OF OMAN By FATMA RASHID HILAL AL-KIYUMI BSc (SQU, Sultanate of Oman) MSc (SCU, Egypt) SUBMITTED IN FULFILMENT OF THE REQUIREMENTS FOR THE DEGREE OF DOCTOR OF PHILOSOPHY UNIVERSITY OF TASMANIA MARCH 2013 DECLARATION OF ORIGINALITY This thesis contains no material which has been accepted for a degree or diploma by the University or any other institution, except by way of background information and duly acknowledged in the thesis. To the best of my knowledge and belief, no material previously published or written by another person except where due acknowledgement is made in the text of the thesis. ………………………… ……………………. Fatma Rashid Al-Kiyumi Date STATEMENT OF ACCESS This thesis may be made available for loan. Copying of any part of this thesis is prohibited for two years from the date this statement was signed; after that time limited copying is permitted in accordance with the Copyright Act 1968. ………………………… ……………………. Fatma Rashid Al-Kiyumi Date Abstract Samples of Arabian pandora Pagellus affinis (Boulenger, 1887) were collected between April 2005 and March 2007, and from April 2008 to March 2009 from two landing sites (Al-Lakbi and Raysut) along the Arabian Sea coast of Oman. The samples were analysed to evaluate the biology and population dynamics of this species. The marginal increment analysis of otoliths showed the formation of one opaque zone and one translucent zone every year. The timing of formation of translucent ring was approximately 4 months (June–September) and the opaque ring formed with the cycle depending on the annual changes in seawater temperature in the Arabian Sea. -

Bennett 2007
CORE Metadata, citation and similar papers at core.ac.uk Provided by South East Academic Libraries System (SEALS) OPTIMISATION OF A SAMPLING PROTOCOL FOR LONG-TERM MONITORING OF TEMPERATE REEF FISHES A thesis submitted in fulfilment of the requirements for the degree of MASTER OF SCIENCE of RHODES UNIVERSITY by RHETT HAMILTON BENNETT June 2007 ABSTRACT Marine Protected Areas (MPAs), the Ecosystem Approach to Fisheries management (EAF) and Integrated Coastal Management (ICM) have been identified as possible alternatives to traditional linefish management measures, which have largely failed. Monitoring and assessment of fish communities on a long-term basis is necessary, and will provide a means to evaluate the effectiveness of such management measures. Therefore, standardised protocols and optimal sampling methods for long-term monitoring (LTM) and assessment of coastal fish communities are essential. This study aimed to identify suitable methods and develop a protocol for assessment of inshore reef fish communities. A suitable location for evaluation of proposed methods was identified in the warm temperate biogeographical region of South Africa, encompassing the well-established Tsitsikamma Coastal National Park MPA and an adjacent exploited area. Chrysoblephus laticeps (roman) was identified as an indicator species for the study, as it has been well-studied and is well represented in the area. Underwater visual census (UVC) and controlled fishing were identified as suitable methods. UVC transects were found to be superior to point counts, in terms of sampling efficiency, variability, bias and required sample size. An effort of two angler hours per fishing station was shown to provide low catch variability, while at the same time a representative catch and low overall cost and required time. -

Review of the Species of the Genus Dentex (Perciformes: Sparidae) in the Western Pacific Defined As the D
Bull. Natl. Mus. Nat. Sci., Ser. A, Suppl. 1, pp. 29–49, March 22, 2007 Review of the Species of the Genus Dentex (Perciformes: Sparidae) in the Western Pacific Defined as the D. hypselosomus complex with the Description of a New Species, Dentex abei and a Redescription of Evynnis tumifrons Yukio Iwatsuki1, Masato Akazaki2 and Nobuhiko Taniguchi3 1 Division of Fisheries Sciences, Faculty of Agriculture, University of Miyazaki, 1–1, Gakuen-kibanadai-nishi, Miyazaki 889–2192, Japan E-mail: [email protected] 2 Deceased on 12 May 1999 3 Laboratory of Applied Population Genetics, Graduate School of Agriculture, Tohoku University, Tutumidori, Amamiya-cho, Aoba-ku, Sendai-City, 981–8555 Japan E-mail: [email protected] Abstract The lectotype of Chrysophrys tumifrons designated by Boeseman (1947) is a specimen of the species currently recognized as Evynnis japonica Tanaka, 1931. The name is, therefore, the senior synonym for that species. Dentex spariformis Ogilby, 1910, based on specimens collected at Moreton Island, Queensland, Australia, is redescribed and resurrected as a valid species, having previously been synonymized with D. tumifrons (Temminck and Schlegel, 1843). Dentex abei sp. nov. is described from ten type and 30 non-type specimens collected in the Ryukyu Islands, Chichi-jima of Ogasawara Islands (ϭBonin Islands), Japan, and Luzon Island, Philippines. These two Dentex species, together with D. hypselosomus Bleeker, 1854 and D. fourmanoiri Akazaki and Séret, 1999 are recognized as a “Dentex hypselosomus complex” and reviewed on the basis of western Pacific and southern Indonesian material. Dentex hypselosomus differs from the others in having three eye-sized fluorescent yellow blotches dorsally, plus a small yellow spot posteriorly on the soft dorsal fin base (vs. -

Hermaphroditism in Fish
Tesis doctoral Evolutionary transitions, environmental correlates and life-history traits associated with the distribution of the different forms of hermaphroditism in fish Susanna Pla Quirante Tesi presentada per a optar al títol de Doctor per la Universitat Autònoma de Barcelona, programa de doctorat en Aqüicultura, del Departament de Biologia Animal, de Biologia Vegetal i Ecologia. Director: Tutor: Dr. Francesc Piferrer Circuns Dr. Lluís Tort Bardolet Departament de Recursos Marins Renovables Departament de Biologia Cel·lular, Institut de Ciències del Mar Fisiologia i Immunologia Consell Superior d’Investigacions Científiques Universitat Autònoma de Barcelona La doctoranda: Susanna Pla Quirante Barcelona, Setembre de 2019 To my mother Agraïments / Acknowledgements / Agradecimientos Vull agrair a totes aquelles persones que han aportat els seus coneixements i dedicació a fer possible aquesta tesi, tant a nivell professional com personal. Per començar, vull agrair al meu director de tesi, el Dr. Francesc Piferrer, per haver-me donat aquesta oportunitat i per haver confiat en mi des del principi. Sempre admiraré i recordaré el teu entusiasme en la ciència i de la contínua formació rebuda, tant a nivell científic com personal. Des del primer dia, a través dels teus consells i coneixements, he experimentat un continu aprenentatge que sens dubte ha derivat a una gran evolució personal. Principalment he après a identificar les meves capacitats i les meves limitacions, i a ser resolutiva davant de qualsevol adversitat. Per tant, el meu més sincer agraïment, que mai oblidaré. During the thesis, I was able to meet incredible people from the scientific world. During my stay at the University of Manchester, where I learned the techniques of phylogenetic analysis, I had one of the best professional experiences with Dr. -

Polysteganus Praeorbitalis (Pisces: Sparidae) to Climate Change in the Agulhas Current System
Modelling the spatial and genetic response of the endemic sparid: Polysteganus praeorbitalis (Pisces: Sparidae) to climate change in the Agulhas Current system A thesis submitted in fulfilment of the requirements for the degree of Master of Science of Rhodes University Grahamstown, South Africa. By: Devin Neil Isemonger November 2013 i Abstract Abstract The Scotsman Seabream, Polysteganus praeorbitalis, is one of several large, slow-growing members of the Sparidae family of fishes endemic to the Agulhas Current system in the Western Indian Ocean (WIO). Relatively little research has been conducted on this species despite its importance to both recreational and commercial line fisheries in South Africa and the drastic decline in catch per unit effort (CPUE) that has been recorded since the 1940s. Changing sea temperatures as a result of global climate change are further expected to affect the distribution and abundance of many fish species based on their thermal tolerances, life histories and population structures. The ability of these species to shift their distribution and adapt to new environments and thermal conditions will depend to some degree on the levels of genetic variation and gene flow, within and between populations. A combined approach using species distribution modelling and genetic analyses may prove to be a useful tool in investigating the potential effects of climate change on the distribution and genetic diversity of species. An ensemble species distribution model (SDM) based on 205 occurrence records and 30 years of Reynolds Optimum Interpolated (OI) sea surface temperature data was constructed to predict the distributional response of P. praeorbitalis to climate change in the Agulhas Current system. -

Chordata Check-List S
1 NEAT (North East Atlantic Taxa): Synoicum Phipps,1774 South Scandinavian marine Chordata Check-List S. pulmonaria (Ellis & Solander,1786) = Amaroucium pomum & = Aplidium ficus Alder & Hancock,1912 compiled at TMBL (Tjärnö Marine Biological Laboratory) by: Öresund-Bohuslän-all Norway-White Sea & Spitsb., Brest-North Sea-Iceland Hans G. Hansson 1989-09-20 / small revisions until March 1996, when it was published as a pdf file on Internet and S. beauchampi (Harant,1927) again republished after small revisions August 1998. Concarneau Citation suggested: Hansson, H.G. (Comp.), NEAT (North East Atlantic Taxa): South Scandinavian marine S. incrustatum (M. Sars,1851) Chordata Check-List. Internet pdf Ed., Aug. 1998. [http://www.tmbl.gu.se]. = Amaroucium incrustratum M. Sars,1851 = Aplidium densum : Picton,1985, non (Giard,1872) Lofoten - Finnmark, Barents Sea, Iceland, N Ireland, Irish Sea Denotations: (™) = Genotype @ = Associated to * = General note S. turgens Phipps,1774 (™) W - NE Norway - Spitsbergen & Bjørnøya N.B.: This is one of several preliminary check-lists, covering S. Scandinavian marine animal (and partly marine Aplidium Savigny,1816 (™ A. lobatum Savigny,1816) protoctist) taxa. Some financial support from (or via) NKMB (Nordiskt Kollegium för Marin Biologi), during the = Amaroucium H. Milne-Edwards,1841 last years of the existence of this organisation (until 1993), is thankfully acknowledged. The primary purpose of = Amaroecium Bronn,1862 these checklists is to facilitate for everyone, trying to identify organisms from the area, to know which species = Amaroeeium Martens, in Jaeger,1880 that earlier have been encountered there, or in neighbouring areas. A secondary purpose is to facilitate for non- = Amaroncium Desmarest, in Chenu,1858 experts to find as correct names as possible for organisms, including names of authors and years of description.