Current and Potential Antiarrhythmic Drugs Targeting Voltage-Gated
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
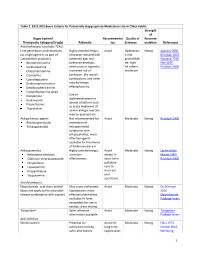
Table 2. 2012 AGS Beers Criteria for Potentially
Table 2. 2012 AGS Beers Criteria for Potentially Inappropriate Medication Use in Older Adults Strength of Organ System/ Recommendat Quality of Recomm Therapeutic Category/Drug(s) Rationale ion Evidence endation References Anticholinergics (excludes TCAs) First-generation antihistamines Highly anticholinergic; Avoid Hydroxyzin Strong Agostini 2001 (as single agent or as part of clearance reduced with e and Boustani 2007 combination products) advanced age, and promethazi Guaiana 2010 Brompheniramine tolerance develops ne: high; Han 2001 Carbinoxamine when used as hypnotic; All others: Rudolph 2008 Chlorpheniramine increased risk of moderate Clemastine confusion, dry mouth, Cyproheptadine constipation, and other Dexbrompheniramine anticholinergic Dexchlorpheniramine effects/toxicity. Diphenhydramine (oral) Doxylamine Use of diphenhydramine in Hydroxyzine special situations such Promethazine as acute treatment of Triprolidine severe allergic reaction may be appropriate. Antiparkinson agents Not recommended for Avoid Moderate Strong Rudolph 2008 Benztropine (oral) prevention of Trihexyphenidyl extrapyramidal symptoms with antipsychotics; more effective agents available for treatment of Parkinson disease. Antispasmodics Highly anticholinergic, Avoid Moderate Strong Lechevallier- Belladonna alkaloids uncertain except in Michel 2005 Clidinium-chlordiazepoxide effectiveness. short-term Rudolph 2008 Dicyclomine palliative Hyoscyamine care to Propantheline decrease Scopolamine oral secretions. Antithrombotics Dipyridamole, oral short-acting* May -

The Pharmacology of Amiodarone and Digoxin As Antiarrhythmic Agents
Part I Anaesthesia Refresher Course – 2017 University of Cape Town The Pharmacology of Amiodarone and Digoxin as Antiarrhythmic Agents Dr Adri Vorster UCT Department of Anaesthesia & Perioperative Medicine The heart contains pacemaker, conduction and contractile tissue. Cardiac arrhythmias are caused by either enhancement or depression of cardiac action potential generation by pacemaker cells, or by abnormal conduction of the action potential. The pharmacological treatment of arrhythmias aims to achieve restoration of a normal rhythm and rate. The resting membrane potential of myocytes is around -90 mV, with the inside of the membrane more negative than the outside. The main extracellular ions are Na+ and Cl−, with K+ the main intracellular ion. The cardiac action potential involves a change in voltage across the cell membrane of myocytes, caused by movement of charged ions across the membrane. This voltage change is triggered by pacemaker cells. The action potential is divided into 5 phases (figure 1). Phase 0: Rapid depolarisation Duration < 2ms Threshold potential must be reached (-70 mV) for propagation to occur Rapid positive charge achieved as a result of increased Na+ conductance through voltage-gated Na+ channels in the cell membrane Phase 1: Partial repolarisation Closure of Na+ channels K+ channels open and close, resulting in brief outflow of K+ and a more negative membrane potential Phase 2: Plateau Duration up to 150 ms Absolute refractory period – prevents further depolarisation and myocardial tetany Result of Ca++ influx -

Tricyclic Antidepressant
Princess Margaret Hospital for Children Emergency Department Guideline PAEDIATRIC ACUTE CARE GUIDELINE Poisoning – Tricyclic Antidepressant Scope (Staff): All Emergency Department Clinicians Scope (Area): Emergency Department This document should be read in conjunction with this DISCLAIMER http://kidshealthwa.com/about/disclaimer/ Poisoning – Tricyclic Antidepressant This guideline is a general approach to tricyclic antidepressant poisoning. For specific details please contact Poisons Information: 131126 or refer to the Toxicology Handbook. Agents: Amitriptyline Clomipramine Dothiepin Doxepin Imipramine Nortriptyline Trimipramine Background Tricyclic antidepressants (TCAs) act on a variety of receptors whose actions include: Noradrenaline reuptake inhibition Central and peripheral anticholinergic effect Fast sodium channel blockade in the myocardium Peripheral alpha1-adrenergic receptor blockade The life threatening effects of acute tricyclic antidepressant (TCA) overdose are: Rapid onset of coma Seizures Cardiac dysrhythmias Page 1 of 6 Emergency Department Guideline Poisoning – Tricyclic Antidepressant Hypotension and central and peripheral anticholinergic effects may also be seen Risk Assessment Most acute accidental paediatric exposures do not result in life threatening toxicity A 10kg child can develop life threatening poisoning with the ingestion of a single tablet (e.g. 150mg amitriptyline) Patients who ingest a large dose of TCA usually develop evidence of intoxication within 2-4 hours, and always within 6 hours If their is suspicion -

Neurochemical Mechanisms Underlying Alcohol Withdrawal
Neurochemical Mechanisms Underlying Alcohol Withdrawal John Littleton, MD, Ph.D. More than 50 years ago, C.K. Himmelsbach first suggested that physiological mechanisms responsible for maintaining a stable state of equilibrium (i.e., homeostasis) in the patient’s body and brain are responsible for drug tolerance and the drug withdrawal syndrome. In the latter case, he suggested that the absence of the drug leaves these same homeostatic mechanisms exposed, leading to the withdrawal syndrome. This theory provides the framework for a majority of neurochemical investigations of the adaptations that occur in alcohol dependence and how these adaptations may precipitate withdrawal. This article examines the Himmelsbach theory and its application to alcohol withdrawal; reviews the animal models being used to study withdrawal; and looks at the postulated neuroadaptations in three systems—the gamma-aminobutyric acid (GABA) neurotransmitter system, the glutamate neurotransmitter system, and the calcium channel system that regulates various processes inside neurons. The role of these neuroadaptations in withdrawal and the clinical implications of this research also are considered. KEY WORDS: AOD withdrawal syndrome; neurochemistry; biochemical mechanism; AOD tolerance; brain; homeostasis; biological AOD dependence; biological AOD use; disorder theory; biological adaptation; animal model; GABA receptors; glutamate receptors; calcium channel; proteins; detoxification; brain damage; disease severity; AODD (alcohol and other drug dependence) relapse; literature review uring the past 25 years research- science models used to study with- of the reasons why advances in basic ers have made rapid progress drawal neurochemistry as well as a research have not yet been translated Din understanding the chemi- reluctance on the part of clinicians to into therapeutic gains and suggests cal activities that occur in the nervous consider new treatments. -

Calcium Current Block by (-)-Pentobarbital, Phenobarbital
The Journal of Neuroscience, August 1993, 13(E): 321 l-3221 Calcium Current Block by (-)-Pentobarbital, Phenobarbital, and CHEB but not (+)-Pentobarbital in Acutely Isolated Hippocampal CA1 Neurons: Comparison with Effects on GABA-activated Cl- Current Jarlath M. H. ffrench-Mullen,’ Jeffery L. Barker,* and Michael A. Rogawski3 ‘Department of Pharmacology, Zeneca Pharmaceuticals Group, Zeneca Inc., Wilmington, Delaware 19897 and *Laboratory of Neurophysiology, and 3Neuronal Excitability Section, Epilepsy Research Branch, National Institute of Neurological Disorders and Stroke, National Institutes of Health, Bethesda, Maryland 20892 Block of a voltage-activated Ca*+ channel current by pheno- Ca*+ current, whereas the sedative effects that occur at high- barbital (PHB), 5-(2-cyclohexylideneethyl)-5-ethyl barbituric er concentrations could reflect stronger Ca2+ current block- acid (CHEB), and the optical R(-)- and S(+)-enantiomers of ade. The powerful sedative-hypnotic action of (-)-PB may pentobarbital (PB) was examined in freshly dissociated adult reflect greater maximal enhancement of GABA responses guinea pig hippocampal CA1 neurons; the effects of the in conjunction with strong inhibition of Ca2+ current. The barbiturates on GABA-activated Cl- current were also char- convulsant action of CHEB is unlikely to be related to its acterized in the same preparation. (-)-PB, PHB, and CHEB effects on the Ca*+ current. produced a reversible, concentration-dependent block of the [Key words: calcium channel, GABA receptor, (-)-pen- peak Ca*+ channel current (3 mM Ba2+ as the charge carrier) tobarbital, (+)-pentobarbital, phenobarbital, CHEB [S-(2-cy- evoked by depolarization from -80 to - 10 mV (I&,, values, clohexylideneethyl)-S-ethyl barbituric acid], CA 1 hippocam- 3.5, 72, and 118 PM, respectively). -
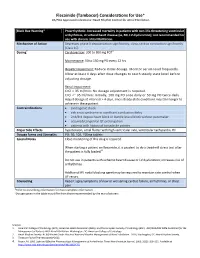
Flecainide Considerations For
Flecainide (Tambocor) Considerations for Use* US/FDA Approved Indications: Heart Rhythm Control for Atrial Fibrillation Black Box Warning* Proarrhythmic. Increased mortality in patients with non-life-threatening ventricular arrhythmias, structural heart disease (ie, MI, LV dysfunction); not recommended for use with chronic atrial fibrillation. Mechanism of Action Depresses phase 0 depolarization significantly, slows cardiac conduction significantly (Class 1C). Dosing† Cardioversion: 200 to 300 mg PO‡1 Maintenance: 50 to 150 mg PO every 12 hrs Hepatic Impairment: Reduce initial dosage. Monitor serum level frequently. Allow at least 4 days after dose changes to reach steady state level before adjusting dosage. Renal Impairment: CrCl > 35 ml/min: No dosage adjustment is required. CrCl <= 35 ml/min: Initially, 100 mg PO once daily or 50 mg PO twice daily. Adjust dosage at intervals > 4 days, since steady-state conditions may take longer to achieve in these patient Contraindications cardiogenic shock sick sinus syndrome or significant conduction delay 2nd/3rd degree heart block or bundle brand block without pacemaker acquired/congenital QT prolongation patients with history of torsade de pointes Major Side Effects hypotension, atrial flutter with high ventricular rate, ventricular tachycardia, HF Dosage forms and Strengths PO: 50, 100, 150mg tablets Special Notes Close monitoring of this drug is required. When starting a patient on flecainide, it is prudent to do a treadmill stress test after the patient is fully loaded.4 Do not use in patients with ischemic heart disease or LV dysfunction; increases risk of arrhythmias. Additional AV nodal blocking agent may be required to maintain rate control when AF recurs. -

A Proposal for the Clinical Use of Flecainide
A Proposal for the Clinical Use of Flecainide JEFFREY L. ANDERSON, MD, JAMES R. STEWART, MD, and BARRY J. CREVEY, MD Effective antiarrhythmic therapy requires a carefully sponse rate is observed in preventing induction of considered approach, including an understanding sustained ventricular tachycardia, and these pa- of the arrhythmia, the underlying cardiac disease tients should be carefully selected. Flecainide is and the drug’s pharmacokinetics. Flecainide is a promising in the treatment of supraventricular new antiarrhythmic drug that may soon be released tachycardias using atrioventricular nodal or extra- for general use. Flecainide demonstrates unsur- nodal reentrant pathways, although this use is still passed efficacy in chronic ventricular arrhythmias investigational in the United States. The drug’s use in stable patients and may become a first-choice for arrhythmias during acute myocardial infarction drug because of its ease of administration, efficacy requires further study. Flecainide possesses modest and favorable tolerance. Twice-daily dosing with negative inotropic potential. Proarrhythmic or other 100 to 200 mg usually provides effective therapy. adverse reactions have occurred primarily in set- Clinical experience suggests flecainide to be indi- tings of high drug level, poor ventricular function cated in the treatment of uniform and multiform or refractory, malignant arrhythmias, suggesting ventricular premature complexes, coupled ven- caution in these groups. tricular premature complexes, and episodes of nonsustained -

Amiodarone (As Hydrochloride)
NEW ZEALAND DATA SHEET ARATAC 1. Product Name Aratac, 100 mg and 200 mg tablet. 2. Qualitative and Quantitative Composition Each Aratac tablet contains 100 mg or 200 mg of amiodarone (as hydrochloride). Aratac tablets contain lactose. For the full list of excipients, see section 6.1. Amiodarone hydrochloride is a fine white crystalline powder. It is slightly soluble in water and is soluble in alcohol and chloroform. It is an amphiphilic compound and contains iodine in its formulation. Each 200 mg tablet of amiodarone contains approximately 75 mg organic iodine. In the steady state, metabolism of 300 mg amiodarone yields 9 mg/day of iodine. 3. Pharmaceutical Form Amiodarone 100 mg Tablet: round, normal convex, white tablet, 8.5 mm diameter, imprinted “AM” | “100” on one side and “G” on the other. Amiodarone 200 mg Tablet: round, normal convex, white tablet, 10.0 mm diameter, imprinted “AM” | “200” on one side and “G” on the other. The tablet can be divided into equal doses. 4. Clinical Particulars 4.1 Therapeutic indications Treatment should be initiated only under hospital or specialist supervision. Tachyarrhythmias associated with Wolff-Parkinson-White Syndrome. Atrial flutter and fibrillation when other agents cannot be used. All types of tachyarrhythmias of paroxysmal nature including: supraventricular, nodal and ventricular tachycardias, ventricular fibrillation; when other agents cannot be used. Tablets are used for stabilisation and long term treatment. 4.2 Dose and method of administration Dose Due to poor absorption and wide inter-patient variability of absorption, the initial loading and subsequent maintenance dosage schedules of the medicine in clinical use has to be individually titrated. -

Guideline for Preoperative Medication Management
Guideline: Preoperative Medication Management Guideline for Preoperative Medication Management Purpose of Guideline: To provide guidance to physicians, advanced practice providers (APPs), pharmacists, and nurses regarding medication management in the preoperative setting. Background: Appropriate perioperative medication management is essential to ensure positive surgical outcomes and prevent medication misadventures.1 Results from a prospective analysis of 1,025 patients admitted to a general surgical unit concluded that patients on at least one medication for a chronic disease are 2.7 times more likely to experience surgical complications compared with those not taking any medications. As the aging population requires more medication use and the availability of various nonprescription medications continues to increase, so does the risk of polypharmacy and the need for perioperative medication guidance.2 There are no well-designed trials to support evidence-based recommendations for perioperative medication management; however, general principles and best practice approaches are available. General considerations for perioperative medication management include a thorough medication history, understanding of the medication pharmacokinetics and potential for withdrawal symptoms, understanding the risks associated with the surgical procedure and the risks of medication discontinuation based on the intended indication. Clinical judgement must be exercised, especially if medication pharmacokinetics are not predictable or there are significant risks associated with inappropriate medication withdrawal (eg, tolerance) or continuation (eg, postsurgical infection).2 Clinical Assessment: Prior to instructing the patient on preoperative medication management, completion of a thorough medication history is recommended – including all information on prescription medications, over-the-counter medications, “as needed” medications, vitamins, supplements, and herbal medications. Allergies should also be verified and documented. -

Angiotensin Modulators/Calcium Channel Blocker Combinations Review 02/05/2009
Angiotensin Modulators/Calcium Channel Blocker Combinations Review 02/05/2009 Copyright © 2004 - 2009 by Provider Synergies, L.L.C. All rights reserved. Printed in the United States of America. All rights reserved. No part of this publication may be reproduced or transmitted in any form or by any means, electronic or mechanical, including photocopying, recording, digital scanning, or via any information storage and retrieval system without the express written consent of Provider Synergies, L.L.C. All requests for permission should be mailed to: Attention: Copyright Administrator Intellectual Property Department Provider Synergies, L.L.C. 5181 Natorp Blvd., Suite 205 Mason, Ohio 45040 The materials contained herein represent the opinions of the collective authors and editors and should not be construed to be the official representation of any professional organization or group, any state Pharmacy and Therapeutics committee, any state Medicaid Agency, or any other clinical committee. This material is not intended to be relied upon as medical advice for specific medical cases and nothing contained herein should be relied upon by any patient, medical professional or layperson seeking information about a specific course of treatment for a specific medical condition. All readers of this material are responsible for independently obtaining medical advice and guidance from their own physician and/or other medical professional in regard to the best course of treatment for their specific medical condition. This publication, inclusive of all forms contained herein, is intended to be educational in nature and is intended to be used for informational purposes only. Comments and suggestions may be sent to [email protected]. -

Treatment for Calcium Channel Blocker Poisoning: a Systematic Review
Clinical Toxicology (2014), 52, 926–944 Copyright © 2014 Informa Healthcare USA, Inc. ISSN: 1556-3650 print / 1556-9519 online DOI: 10.3109/15563650.2014.965827 REVIEW ARTICLE Treatment for calcium channel blocker poisoning: A systematic review M. ST-ONGE , 1,2,3 P.-A. DUB É , 4,5,6 S. GOSSELIN ,7,8,9 C. GUIMONT , 10 J. GODWIN , 1,3 P. M. ARCHAMBAULT , 11,12,13,14 J.-M. CHAUNY , 15,16 A. J. FRENETTE , 15,17 M. DARVEAU , 18 N. LE SAGE , 10,14 J. POITRAS , 11,12 J. PROVENCHER , 19 D. N. JUURLINK , 1,20,21 and R. BLAIS 7 1 Ontario and Manitoba Poison Centre, Toronto, ON, Canada 2 Institute of Medical Science, University of Toronto, Toronto, ON, Canada 3 Department of Clinical Pharmacology and Toxicology, University of Toronto, Toronto, ON, Canada 4 Direction of Environmental Health and Toxicology, Institut national de sant é publique du Qu é bec, Qu é bec, QC, Canada 5 Centre Hospitalier Universitaire de Qu é bec, Qu é bec, QC, Canada 6 Faculty of Pharmacy, Université Laval, Qu é bec, QC, Canada 7 Centre antipoison du Qu é bec, Qu é bec, QC, Canada 8 Department of Medicine, McGill University, Montr é al, QC, Canada 9 Toxicology Consulting Service, McGill University Health Centre, Montr é al, QC, Canada 10 Centre Hospitalier Universitaire de Qu é bec, Qu é bec, QC, Canada 11 Centre de sant é et services sociaux Alphonse-Desjardins (CHAU de Lévis), L é vis, QC, Canada 12 Department of Family Medicine and Emergency Medicine, Universit é Laval, Québec, QC, Canada 13 Division de soins intensifs, Universit é Laval, Qu é bec, QC, Canada 14 Populations -

Amiodarone Enhances Anticonvulsive Effect of Oxcarbazepine and Pregabalin in the Mouse Maximal Electroshock Model
International Journal of Molecular Sciences Article Amiodarone Enhances Anticonvulsive Effect of Oxcarbazepine and Pregabalin in the Mouse Maximal Electroshock Model Monika Banach 1 , Monika Rudkowska 1, Agata Sumara 2 and Kinga Borowicz-Reutt 1,* 1 Independent Unit of Experimental Neuropathophysiology, Medical University of Lublin, Jaczewskiego 8b, PL-20-090 Lublin, Poland; [email protected] (M.B.); [email protected] (M.R.) 2 Department of Pathophysiology, Medical University of Lublin, Jaczewskiego 8b, PL-20-090 Lublin, Poland; [email protected] * Correspondence: [email protected] Abstract: Accumulating experimental studies show that antiarrhythmic and antiepileptic drugs share some molecular mechanisms of action and can interact with each other. In this study, the influence of amiodarone (a class III antiarrhythmic drug) on the antiseizure action of four second-generation antiepileptic drugs was evaluated in the maximal electroshock model in mice. Amiodarone, although ineffective in the electroconvulsive threshold test, significantly potentiated the antielectroshock activ- ity of oxcarbazepine and pregabalin. Amiodarone, given alone or in combination with oxcarbazepine, lamotrigine, or topiramate, significantly disturbed long-term memory in the passive-avoidance task in mice. Brain concentrations of antiepileptic drugs were not affected by amiodarone. However, the brain concentration of amiodarone was significantly elevated by oxcarbazepine, topiramate, and pregabalin. Additionally, oxcarbazepine and pregabalin elevated the brain concentration of desethylamiodarone, the main metabolite of amiodarone. In conclusion, potentially beneficial action of amiodarone in epilepsy patients seems to be limited by neurotoxic effects of amiodarone. Although results of this study should still be confirmed in chronic protocols of treatment, special precautions Citation: Banach, M.; Rudkowska, are recommended in clinical conditions.