Morphs Salvelinus Alpinus
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Domain of Escherichia Coli 16S Ribosomal RNA Using Site-Directed Photoaffinity Crosslinking
Downloaded from rnajournal.cshlp.org on October 6, 2021 - Published by Cold Spring Harbor Laboratory Press RNA (1998), 4:1455–1466+ Cambridge University Press+ Printed in the USA+ Copyright © 1998 RNA Society+ Analysis of the conformation of the 39 major domain of Escherichia coli 16S ribosomal RNA using site-directed photoaffinity crosslinking ALEXANDRE MONTPETIT,1 CATHERINE PAYANT,1 JAMES M. NOLAN,2 and LÉA BRAKIER-GINGRAS1 1Département de Biochimie, Université de Montréal, Montréal, Québec H3T 1J4, Canada 2Department of Biochemistry, Tulane University Medical Center, New Orleans, Louisiana 70112, USA ABSTRACT The 39 major domain of Escherichia coli 16S rRNA, which occupies the head of the small ribosomal subunit, is involved in several functions of the ribosome. We have used a site-specific crosslinking procedure to gain further insights into the higher-order structure of this domain. Circularly permuted RNAs were used to introduce an azi- dophenacyl group at specific positions within the 39 major domain. Crosslinks were generated in a high-ionic strength buffer that has been used for ribosome reconstitution studies and so enables the RNA to adopt a structure recognized by ribosomal proteins. The crosslinking sites were identified by primer extension and confirmed by assessing the mobility of the crosslinked RNA lariats in denaturing polyacrylamide gels. Eight crosslinks were characterized. Among them, one crosslink demonstrates that helix 28 is proximal to the top of helix 34, and two others show that the 1337 region, located in an internal loop at the junction of helices 29, 30, 41, and 42, is proximal to the center of helix 30 and to a segment connecting helix 28 to helix 29. -

The Microbiota-Produced N-Formyl Peptide Fmlf Promotes Obesity-Induced Glucose
Page 1 of 230 Diabetes Title: The microbiota-produced N-formyl peptide fMLF promotes obesity-induced glucose intolerance Joshua Wollam1, Matthew Riopel1, Yong-Jiang Xu1,2, Andrew M. F. Johnson1, Jachelle M. Ofrecio1, Wei Ying1, Dalila El Ouarrat1, Luisa S. Chan3, Andrew W. Han3, Nadir A. Mahmood3, Caitlin N. Ryan3, Yun Sok Lee1, Jeramie D. Watrous1,2, Mahendra D. Chordia4, Dongfeng Pan4, Mohit Jain1,2, Jerrold M. Olefsky1 * Affiliations: 1 Division of Endocrinology & Metabolism, Department of Medicine, University of California, San Diego, La Jolla, California, USA. 2 Department of Pharmacology, University of California, San Diego, La Jolla, California, USA. 3 Second Genome, Inc., South San Francisco, California, USA. 4 Department of Radiology and Medical Imaging, University of Virginia, Charlottesville, VA, USA. * Correspondence to: 858-534-2230, [email protected] Word Count: 4749 Figures: 6 Supplemental Figures: 11 Supplemental Tables: 5 1 Diabetes Publish Ahead of Print, published online April 22, 2019 Diabetes Page 2 of 230 ABSTRACT The composition of the gastrointestinal (GI) microbiota and associated metabolites changes dramatically with diet and the development of obesity. Although many correlations have been described, specific mechanistic links between these changes and glucose homeostasis remain to be defined. Here we show that blood and intestinal levels of the microbiota-produced N-formyl peptide, formyl-methionyl-leucyl-phenylalanine (fMLF), are elevated in high fat diet (HFD)- induced obese mice. Genetic or pharmacological inhibition of the N-formyl peptide receptor Fpr1 leads to increased insulin levels and improved glucose tolerance, dependent upon glucagon- like peptide-1 (GLP-1). Obese Fpr1-knockout (Fpr1-KO) mice also display an altered microbiome, exemplifying the dynamic relationship between host metabolism and microbiota. -

Intimate Relations—Mitochondria and Ageing
International Journal of Molecular Sciences Review Intimate Relations—Mitochondria and Ageing Michael Webb and Dionisia P. Sideris * Mitobridge Inc., an Astellas Company, 1030 Massachusetts Ave, Cambridge, MA 02138, USA; [email protected] * Correspondence: [email protected] Received: 29 August 2020; Accepted: 6 October 2020; Published: 14 October 2020 Abstract: Mitochondrial dysfunction is associated with ageing, but the detailed causal relationship between the two is still unclear. Wereview the major phenomenological manifestations of mitochondrial age-related dysfunction including biochemical, regulatory and energetic features. We conclude that the complexity of these processes and their inter-relationships are still not fully understood and at this point it seems unlikely that a single linear cause and effect relationship between any specific aspect of mitochondrial biology and ageing can be established in either direction. Keywords: mitochondria; ageing; energetics; ROS; gene regulation 1. Introduction The last two decades have witnessed a dramatic transformation in our view of mitochondria, their basic biology and functions. While still regarded as functioning primarily as the eukaryotic cell’s generator of energy in the form of adenosine triphosphate (ATP) and nicotinamide adenine dinucleotide (reduced form; NADH), mitochondria are now recognized as having a plethora of functions, including control of apoptosis, regulation of calcium, forming a signaling hub and the synthesis of various bioactive molecules. Their biochemical functions beyond ATP supply include biosynthesis of lipids and amino acids, formation of iron sulphur complexes and some stages of haem biosynthesis and the urea cycle. They exist as a dynamic network of organelles that under normal circumstances undergo a constant series of fission and fusion events in which structural, functional and encoding (mtDNA) elements are subject to redistribution throughout the network. -
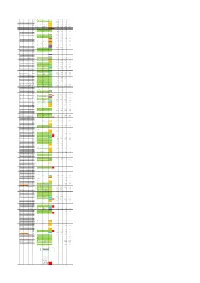
End Strand of Gene Gene Name Gene Function Starnd of Trascript
Genomic TSS strand Gene starnd of Transcipt relatively to TSS Start End Gene name coordinates relatively TSS type Control_fwd TEX_fwd Control_rev TEX_rev of gene function trascript gene group of TSS to ATG ribosomal 93 854 + rps12 + 1 protein S12 1 1 14.0638317 20.7539794 0 0 93 324 exon 1 + 111 rps12 intron 1 1 13.90756687 18.07149224 0.781323982 0.423550599 829 854 exon 2 + 496 rps12 exon2 2 24.22104343 15.24782157 0 0 30S 904 1371 + gene rps7 ribosomal + 2 protein S7 1303 -209 rps7 inter (ndhB) 2 20.47068832 9.035746118 0.625059185 0.847101199 NADH dehydrogen 1512 3535 + gene ndhB (ndh2) + ase subunit 3 2 1696 ndhB exon 1-inter 2 3.594090315 2.964854195 0.468794389 0.282367066 + 2209 ndhB exon 1-inter 2 46.09811492 4.09432246 0.468794389 0.423550599 1512 2237 exon 1 + 2756 ndhB exon 2 -inter 2 43.28534858 4.800240125 0.312529593 0.282367066 2756 3535 exon 2 + 3090 ndhB exon 2 -inter 2 17.50165719 15.95373924 0.312529593 0 + 3192 ndhB exon 2 -inter 2 140.6383167 117.6058831 2.812766334 1.694202397 - 3462 ndhB exon 2 -inter 3 1.406383167 1.129468265 1.406383167 3.67077186 4 3633 3712 + tRNA tRNA-Leu-CAA + 3610 -23 tRNA-Leu 1 77.19480938 84.85130339 0.625059185 0 + 3633 1 tRNA-Leu 1 359.5652963 649.0207016 0.781323982 0 photosyste 3954 4058 + gene psbM m II protein + 5 M 3775 -179 psbM 2 20.47068832 12.00060031 0 0.141183533 + 3954 psbM-0 2 69.22530477 28.37789015 0.156264796 0 hypothetical 4182 5073 + gene ycf66 6 protein 4182 4287 exon 1 4772 5073 exon 2 7 5202 5113 - gene ycf (ORF29) - 5299 orf29 inter 1 0 0 3.125295926 3.67077186 -

WO 2016/200987 Al 15 December 2016 (15.12.2016) P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2016/200987 Al 15 December 2016 (15.12.2016) P O P C T (51) International Patent Classification: Trudi, A.; Indigo Agriculture, Inc., 500 Rutherford Aven A01N 63/02 (2006.01) A01C 1/00 (2006.01) ue, North Building, Boston, MA 02129 (US). A01N 63/00 (2006.01) A01C 1/06 (2006.01) (74) Agents: HUBL, Susan, T. et al; Fenwick & West LLP, (21) International Application Number: Silicon Valley Center, 801 California Street, Mountain PCT/US20 16/036504 View, CA 94041 (US). (22) International Filing Date: (81) Designated States (unless otherwise indicated, for every 8 June 2016 (08.06.2016) kind of national protection available): AE, AG, AL, AM, AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, (25) Filing Language: English BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, (26) Publication Language: English DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, HN, HR, HU, ID, IL, N , IR, IS, JP, KE, KG, KN, KP, KR, (30) Priority Data: KZ, LA, LC, LK, LR, LS, LU, LY, MA, MD, ME, MG, 62/172,748 8 June 2015 (08.06.2015) US MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, OM, 62/172,750 8 June 2015 (08.06.2015) US PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, SC, 62/172,755 8 June 2015 (08.06.2015) us SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, 62/3 16,386 31 March 2016 (3 1.03.2016) us TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. -

Transcriptomic Regulation of Alternative Phenotypic Trajectories in Embryos of the Annual Killifish Austrofundulus Limnaeus
Portland State University PDXScholar Dissertations and Theses Dissertations and Theses Fall 11-30-2017 Transcriptomic Regulation of Alternative Phenotypic Trajectories in Embryos of the Annual Killifish Austrofundulus limnaeus Amie L. Romney Portland State University Follow this and additional works at: https://pdxscholar.library.pdx.edu/open_access_etds Part of the Biology Commons, and the Genetics and Genomics Commons Let us know how access to this document benefits ou.y Recommended Citation Romney, Amie L., "Transcriptomic Regulation of Alternative Phenotypic Trajectories in Embryos of the Annual Killifish Austrofundulus limnaeus" (2017). Dissertations and Theses. Paper 4033. https://doi.org/10.15760/etd.5917 This Dissertation is brought to you for free and open access. It has been accepted for inclusion in Dissertations and Theses by an authorized administrator of PDXScholar. Please contact us if we can make this document more accessible: [email protected]. Transcriptomic Regulation of Alternative Phenotypic Trajectories in embryos of the Annual Killifish Austrofundulus limnaeus by Amie Lynn Thomas Romney A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in Biology Dissertation Committee Jason Podrabsky, Chair Suzanne Estes Bradley Buckley Todd Rosenstiel Dirk Iwata-Reuyl Portland State University 2017 © 2017 Amie Lynn Thomas Romney ABSTRACT The Annual Killifish, Austrofundulus limnaeus, survives the seasonal drying of their pond habitat in the form of embryos entering diapause midway through development. The diapause trajectory is one of two developmental phenotypes. Alternatively, individuals can “escape” entry into diapause and develop continuously until hatching. The alternative phenotypes of A. limnaeus are a form of developmental plasticity that provides this species with a physiological adaption for surviving stressful environments. -

Open FINAL GRAD SCHOOL.Pdf
The Pennsylvania State University The Graduate School Eberly College of Science PHOSPHOPROTEOMIC ANALYSIS OF RIBOSOMAL PROTEINS: IMPLICATIONS IN TRANSLATION AND APOPTOSIS A Dissertation in Biochemistry, Microbiology, and Molecular Biology by Jennifer Lynn Miller © 2009 Jennifer Lynn Miller Submitted in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy May 2009 The dissertation of Jennifer Lynn Miller was reviewed and approved* by the following: Emine C. Koc Assistant Professor Biochemistry and Molecular Biology Dissertation Advisor Chair of Committee Robert A. Schlegel Professor of Biochemistry and Molecular Biology Wendy Hanna-Rose Assistant Professor Biochemistry and Molecular Biology Ming Tien Professor of Biochemistry Erin D. Sheets Assistant Professor of Chemistry Richard J. Frisque Professor of Molecular Virology Head of the Department of Biochemistry and Molecular Biology *Signatures are on file in the Graduate School. ABSTRACT Mammalian mitochondrial ribosomes synthesize thirteen proteins that are essential for oxidative phosphorylation. Besides having a major role in ATP synthesis, mitochondria also contribute to biochemical processes coordinating apoptosis, mitochondrial diseases, and aging in eukaryotic cells. This unique class of ribosomes is protein-rich and distinct from cytoplasmic ribosomes. However, mitochondrial ribosomes (55S) share a significant homology to bacterial ribosomes (70S), particularly in size, the general mechanism of translation, and ribosomal protein content. Due to the overall resemblance between the two systems and the earlier reports of post-translational modifications, we investigated how phosphorylation of ribosomal proteins from bacteria and mitochondria regulates translation and other acquired roles. Identification of twenty- four phosphorylated 70S and 55S ribosomal proteins as well as the potential endogenous kinase was achieved using 2D-gel electrophoresis and tandem mass spectrometry. -

S6:S18 Ribosomal Protein Complex Interacts with a Structural Motif Present in Its Own Mrna
Downloaded from rnajournal.cshlp.org on September 28, 2021 - Published by Cold Spring Harbor Laboratory Press BIOINFORMATICS S6:S18 ribosomal protein complex interacts with a structural motif present in its own mRNA DOROTA MATELSKA,1 ELZBIETA PURTA,1 SYLWIA PANEK,1 MICHAL J. BONIECKI,1 JANUSZ M. BUJNICKI,1,2,3 and STANISLAW DUNIN-HORKAWICZ1,3 1Laboratory of Bioinformatics and Protein Engineering, International Institute of Molecular and Cell Biology, Warsaw, 02-109, Poland 2Bioinformatics Laboratory, Institute of Molecular Biology and Biotechnology, Faculty of Biology, Adam Mickiewicz University, Poznan´,61-614, Poland ABSTRACT Prokaryotic ribosomal protein genes are typically grouped within highly conserved operons. In many cases, one or more of the encoded proteins not only bind to a specific site in the ribosomal RNA, but also to a motif localized within their own mRNA, and thereby regulate expression of the operon. In this study, we computationally predicted an RNA motif present in many bacterial phyla within the 5′ untranslated region of operons encoding ribosomal proteins S6 and S18. We demonstrated that the S6:S18 complex binds to this motif, which we hereafter refer to as the S6:S18 complex-binding motif (S6S18CBM). This motif is a conserved CCG sequence presented in a bulge flanked by a stem and a hairpin structure. A similar structure containing a CCG trinucleotide forms the S6:S18 complex binding site in 16S ribosomal RNA. We have constructed a 3D structural model of a S6:S18 complex with S6S18CBM, which suggests that the CCG trinucleotide in a specific structural context may be specifically recognized by the S18 protein. -

The Genome of Austrofundulus Limnaeus Offers Insights Into Extreme Vertebrate Stress Tolerance and Embryonic Development Josiah T
Wagner et al. BMC Genomics (2018) 19:155 https://doi.org/10.1186/s12864-018-4539-7 RESEARCH ARTICLE Open Access The genome of Austrofundulus limnaeus offers insights into extreme vertebrate stress tolerance and embryonic development Josiah T. Wagner1,5*, Param Priya Singh2, Amie L. Romney1, Claire L. Riggs1, Patrick Minx3, Steven C. Woll1, Jake Roush1, Wesley C. Warren3, Anne Brunet2,4 and Jason E. Podrabsky1 Abstract Background: The annual killifish Austrofundulus limnaeus inhabits ephemeral ponds in northern Venezuela, South America, and is an emerging extremophile model for vertebrate diapause, stress tolerance, and evolution. Embryos of A. limnaeus regularly experience extended periods of desiccation and anoxia as a part of their natural history and have unique metabolic and developmental adaptations. Currently, there are limited genomic resources available for gene expression and evolutionary studies that can take advantage of A. limnaeus as a unique model system. Results: We describe the first draft genome sequence of A. limnaeus. The genome was assembled de novo using a merged assembly strategy and was annotated using the NCBI Eukaryotic Annotation Pipeline. We show that the assembled genome has a high degree of completeness in genic regions that is on par with several other teleost genomes. Using RNA-seq and phylogenetic-based approaches, we identify several candidate genes that may be important for embryonic stress tolerance and post-diapause development in A. limnaeus. Several of these genes include heat shock proteins that have unique expression patterns in A. limnaeus embryos and at least one of these may be under positive selection. Conclusion: The A. limnaeus genome is the first South American annual killifish genome made publicly available. -

Evolution of Translation the Ribosome
University of Illinois at Urbana-Champaign Luthey-Schulten Group NIH Resource for Macromolecular Modeling and Bioinformatics Computational Biophysics Workshop Evolution of Translation The Ribosome VMD Developer: John Stone MultiSeq Developers Tutorial Authors Elijah Roberts Ke Chen John Eargle John Eargle Dan Wright Tyler Earnest Jonathan Lai Zan Luthey-Schulten April 2015 A current version of this tutorial is available at http://www.scs.illinois.edu/~schulten/tutorials/ribosome CONTENTS 2 Contents Introduction 3 Requirements . 4 1 The Ribosomal SSU and associated structures: [30 minutes] 4 2 The Ribosome LSU and associated structures: [30 minutes] 9 2.1 The peptidyl-transferase center . 10 3 Ribosome Origins: [30 minutes] 11 3.1 Hypothesis on the evolution of the ribosome . 11 4 Ribosomal signatures: [60 minutes] 12 4.1 Definition and classification of the ribosomal signatures . 14 4.2 Contribution of ribosomal signatures to phylogenetic separation . 17 4.3 Functional roles of signatures in ribosomal assembly . 20 5 Kinetic Model of Ribosome assembly: [30 minutes] 22 Acknowledgements 26 CONTENTS 3 Introduction The ribosome is a large structure found in all living cells that serves as the main translation machinery of the cell. Messenger RNA (mRNA), transcribed from the organism's genome, binds with the ribosome to commence translation to protein. As explained in the previous tutorials [1, 2, 3], many other cellular components, including tRNA, the aminoacyl-tRNA synthetases, and the elonga- tion factors participate in the translation process; however, the ribosome is the central machinery that assembles a protein from a transcribed gene. Solving the structure of the ribosome was awarded the Nobel Prize in Chemistry in 2009 [4]. -

The Presence of Highly Disruptive 16S Rrna Mutations in Clinical Samples Indicates a Wider Role for Mutations of the Mitochondrial Ribosome in Human Disease
Elson JL, Smith PM, Greaves LC, Lightowlers RN, Chrzanowska-Lightowlers ZMA, Taylor RW, Vila-Sanjurjo A. The presence of highly disruptive 16S rRNA mutations in clinical samples indicates a wider role for mutations of the mitochondrial ribosome in human disease. Mitochondrion 2015, 25, 17-27. Copyright: © 2015 The Authors. Elsevier B.V. and Mitochondria Research Society. This is an open access article under the CC BY license (http://creativecommons.org/licenses/by/4.0/). DOI link to article: http://dx.doi.org/10.1016/j.mito.2015.08.004 Date deposited: 02/10/2015 This work is licensed under a Creative Commons Attribution 4.0 International License Newcastle University ePrints - eprint.ncl.ac.uk Mitochondrion 25 (2015) 17–27 Contents lists available at ScienceDirect Mitochondrion journal homepage: www.elsevier.com/locate/mito The presence of highly disruptive 16S rRNA mutations in clinical samples indicates a wider role for mutations of the mitochondrial ribosome in human disease Joanna L. Elson a,b,1, Paul M. Smith c,d,1, Laura C. Greaves d, Robert N. Lightowlers e, Zofia M.A. Chrzanowska-Lightowlers d,RobertW.Taylord, Antón Vila-Sanjurjo f,⁎ a Institute of Genetic Medicine, Newcastle University, Newcastle upon Tyne NE1 3BZ, United Kingdom b Centre for Human Metabonomics, North-West University, Potchefstroom, South Africa c Institute of Medical Sciences, Ninewells Hospital and Medical School, Dundee University, Dundee DD1 9SY, Scotland, UK d Wellcome Trust Centre for Mitochondrial Research, Institute of Neuroscience, Newcastle University, -

Mitoriboscins: Mitochondrial-Based Therapeutics Targeting Cancer Stem Cells (Cscs), Bacteria and Pathogenic Yeast
www.impactjournals.com/oncotarget/ Oncotarget, 2017, Vol. 8, (No. 40), pp: 67457-67472 Research Paper Mitoriboscins: Mitochondrial-based therapeutics targeting cancer stem cells (CSCs), bacteria and pathogenic yeast Bela Ozsvari1,2, Marco Fiorillo1,2,3, Gloria Bonuccelli1,2, Anna Rita Cappello3, Luca Frattaruolo3, Federica Sotgia1,2, Rachel Trowbridge5, Richard Foster4,5 and Michael P. Lisanti1,2 1 Translational Medicine, School of Environment & Life Sciences, University of Salford, Greater Manchester, UK 2 The Paterson Institute, University of Manchester, Withington, UK 3 The Department of Pharmacy, Health and Nutritional Sciences, The University of Calabria, Cosenza, Italy 4 Astbury Centre for Structural Molecular Biology, University of Leeds, West Yorkshire, UK 5 School of Chemistry, Faculty of Mathematics and Physical Sciences, University of Leeds, West Yorkshire, UK Correspondence to: Michael P. Lisanti, email: [email protected] Correspondence to: Richard Foster, email: [email protected] Keywords: antibiotic, drug design, mitochondrial ribosome, mitoribosome, mitochondria Received: April 02, 2017 Accepted: May 17, 2017 Published: July 07, 2017 Copyright: Ozsvari et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC-BY), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. ABSTRACT The “endo-symbiotic theory of mitochondrial evolution” states that mitochondrial organelles evolved from engulfed aerobic bacteria, after millions of years of symbiosis and adaptation. Here, we have exploited this premise to design new antibiotics and novel anti-cancer therapies, using a convergent approach. First, virtual high- throughput screening (vHTS) and computational chemistry were used to identify novel compounds binding to the 3D structure of the mammalian mitochondrial ribosome.