Epitranscriptomics of Mammalian Mitochondrial Ribosomal RNA
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Case Report Clinical Report of a 17Q12 Microdeletion with Additionally Unreported Clinical Features
Hindawi Publishing Corporation Case Reports in Genetics Volume 2014, Article ID 264947, 6 pages http://dx.doi.org/10.1155/2014/264947 Case Report Clinical Report of a 17q12 Microdeletion with Additionally Unreported Clinical Features Jennifer L. Roberts,1 Stephanie K. Gandomi,2 Melissa Parra,2 Ira Lu,2 Chia-Ling Gau,2 Majed Dasouki,1,3 and Merlin G. Butler1 1 The University of Kansas Medical Center, Kansas City, KS 66160, USA 2 Ambry Genetics, Aliso Viejo, CA 92656, USA 3 King Faisal Specialist Hospital and Research Center, Riyadh 12713, Saudi Arabia Correspondence should be addressed to Stephanie K. Gandomi; [email protected] Received 18 March 2014; Revised 13 May 2014; Accepted 14 May 2014; Published 2 June 2014 Academic Editor: Philip D. Cotter Copyright © 2014 Jennifer L. Roberts et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Copy number variations involving the 17q12 region have been associated with developmental and speech delay, autism, aggression, self-injury, biting and hitting, oppositional defiance, inappropriate language, and auditory hallucinations. We present a tall- appearing 17-year-old boy with marfanoid habitus, hypermobile joints, mild scoliosis, pectus deformity, widely spaced nipples, pes cavus, autism spectrum disorder, intellectual disability, and psychiatric manifestations including physical and verbal aggression, obsessive-compulsive behaviors, and oppositional defiance. An echocardiogram showed borderline increased aortic root size. An abdominal ultrasound revealed a small pancreas, mild splenomegaly with a 1.3 cm accessory splenule, and normal kidneys and liver. -

Analysis of Gene Expression Data for Gene Ontology
ANALYSIS OF GENE EXPRESSION DATA FOR GENE ONTOLOGY BASED PROTEIN FUNCTION PREDICTION A Thesis Presented to The Graduate Faculty of The University of Akron In Partial Fulfillment of the Requirements for the Degree Master of Science Robert Daniel Macholan May 2011 ANALYSIS OF GENE EXPRESSION DATA FOR GENE ONTOLOGY BASED PROTEIN FUNCTION PREDICTION Robert Daniel Macholan Thesis Approved: Accepted: _______________________________ _______________________________ Advisor Department Chair Dr. Zhong-Hui Duan Dr. Chien-Chung Chan _______________________________ _______________________________ Committee Member Dean of the College Dr. Chien-Chung Chan Dr. Chand K. Midha _______________________________ _______________________________ Committee Member Dean of the Graduate School Dr. Yingcai Xiao Dr. George R. Newkome _______________________________ Date ii ABSTRACT A tremendous increase in genomic data has encouraged biologists to turn to bioinformatics in order to assist in its interpretation and processing. One of the present challenges that need to be overcome in order to understand this data more completely is the development of a reliable method to accurately predict the function of a protein from its genomic information. This study focuses on developing an effective algorithm for protein function prediction. The algorithm is based on proteins that have similar expression patterns. The similarity of the expression data is determined using a novel measure, the slope matrix. The slope matrix introduces a normalized method for the comparison of expression levels throughout a proteome. The algorithm is tested using real microarray gene expression data. Their functions are characterized using gene ontology annotations. The results of the case study indicate the protein function prediction algorithm developed is comparable to the prediction algorithms that are based on the annotations of homologous proteins. -

Domain of Escherichia Coli 16S Ribosomal RNA Using Site-Directed Photoaffinity Crosslinking
Downloaded from rnajournal.cshlp.org on October 6, 2021 - Published by Cold Spring Harbor Laboratory Press RNA (1998), 4:1455–1466+ Cambridge University Press+ Printed in the USA+ Copyright © 1998 RNA Society+ Analysis of the conformation of the 39 major domain of Escherichia coli 16S ribosomal RNA using site-directed photoaffinity crosslinking ALEXANDRE MONTPETIT,1 CATHERINE PAYANT,1 JAMES M. NOLAN,2 and LÉA BRAKIER-GINGRAS1 1Département de Biochimie, Université de Montréal, Montréal, Québec H3T 1J4, Canada 2Department of Biochemistry, Tulane University Medical Center, New Orleans, Louisiana 70112, USA ABSTRACT The 39 major domain of Escherichia coli 16S rRNA, which occupies the head of the small ribosomal subunit, is involved in several functions of the ribosome. We have used a site-specific crosslinking procedure to gain further insights into the higher-order structure of this domain. Circularly permuted RNAs were used to introduce an azi- dophenacyl group at specific positions within the 39 major domain. Crosslinks were generated in a high-ionic strength buffer that has been used for ribosome reconstitution studies and so enables the RNA to adopt a structure recognized by ribosomal proteins. The crosslinking sites were identified by primer extension and confirmed by assessing the mobility of the crosslinked RNA lariats in denaturing polyacrylamide gels. Eight crosslinks were characterized. Among them, one crosslink demonstrates that helix 28 is proximal to the top of helix 34, and two others show that the 1337 region, located in an internal loop at the junction of helices 29, 30, 41, and 42, is proximal to the center of helix 30 and to a segment connecting helix 28 to helix 29. -

Nucleosome Positioning Signals in Genomic DNA
Downloaded from genome.cshlp.org on September 26, 2021 - Published by Cold Spring Harbor Laboratory Press Letter Nucleosome positioning signals in genomic DNA Heather E. Peckham,1,2 Robert E. Thurman,3 Yutao Fu,1 John A. Stamatoyannopoulos,4 William Stafford Noble,4,5 Kevin Struhl,6 and Zhiping Weng1,2,7 1Bioinformatics Program, Boston University, Boston, Massachusetts 02215, USA; 2Department of Biomedical Engineering, Boston University, Boston, Massachusetts 02215, USA; 3Division of Medical Genetics, University of Washington, Seattle, Washington 98195, USA; 4Department of Genome Sciences, University of Washington, Seattle, Washington 98195, USA; 5Department of Computer Science and Engineering, University of Washington, Seattle, Washington 98195, USA; 6Department of Biological Chemistry and Molecular Pharmacology, Harvard Medical School, Boston, Massachusetts 02115, USA Although histones can form nucleosomes on virtually any genomic sequence, DNA sequences show considerable variability in their binding affinity. We have used DNA sequences of Saccharomyces cerevisiae whose nucleosome binding affinities have been experimentally determined (Yuan et al. 2005) to train a support vector machine to identify the nucleosome formation potential of any given sequence of DNA. The DNA sequences whose nucleosome formation potential are most accurately predicted are those that contain strong nucleosome forming or inhibiting signals and are found within nucleosome length stretches of genomic DNA with continuous nucleosome formation or inhibition signals. We have accurately predicted the experimentally determined nucleosome positions across a well-characterized promoter region of S. cerevisiae and identified strong periodicity within 199 center-aligned mononucleosomes studied recently (Segal et al. 2006) despite there being no periodicity information used to train the support vector machine. -

Targeted Exome Sequencing Provided Comprehensive Genetic Diagnosis of Congenital Anomalies of the Kidney and Urinary Tract
Journal of Clinical Medicine Article Targeted Exome Sequencing Provided Comprehensive Genetic Diagnosis of Congenital Anomalies of the Kidney and Urinary Tract 1,2, 3,4, 3 1,5 Yo Han Ahn y, Chung Lee y, Nayoung K. D. Kim , Eujin Park , Hee Gyung Kang 1,2,6,* , Il-Soo Ha 1,2,6, Woong-Yang Park 3,4,7 and Hae Il Cheong 1,2,6 1 Department of Pediatrics, Seoul National University College of Medicine, Seoul 03080, Korea; [email protected] (Y.H.A.); [email protected] (E.P.); [email protected] (I.-S.H.); [email protected] (H.I.C.) 2 Department of Pediatrics, Seoul National University Children’s Hospital, Seoul 03080, Korea 3 Samsung Genome Institute, Samsung Medical Center, Seoul 06351, Korea; [email protected] (C.L.); [email protected] (N.K.D.K.); [email protected] (W.-Y.P.) 4 Department of Health Sciences and Technology, Samsung Advanced Institute for Health Sciences and Technology, Sungkyunkwan University, Seoul 06351, Korea 5 Department of Pediatrics, Kangnam Sacred Heart Hospital, Hallym University College of Medicine, Seoul 07441, Korea 6 Kidney Research Institute, Medical Research Center, Seoul National University College of Medicine, Seoul 03080, Korea 7 Department of Molecular Cell Biology, Sungkyunkwan University School of Medicine, Suwon 16419, Korea * Correspondence: [email protected] These authors equally contributed to this article. y Received: 31 January 2020; Accepted: 8 March 2020; Published: 10 March 2020 Abstract: Congenital anomalies of the kidney and urinary tract (CAKUT) are the most common cause of chronic kidney disease in children. -

The Microbiota-Produced N-Formyl Peptide Fmlf Promotes Obesity-Induced Glucose
Page 1 of 230 Diabetes Title: The microbiota-produced N-formyl peptide fMLF promotes obesity-induced glucose intolerance Joshua Wollam1, Matthew Riopel1, Yong-Jiang Xu1,2, Andrew M. F. Johnson1, Jachelle M. Ofrecio1, Wei Ying1, Dalila El Ouarrat1, Luisa S. Chan3, Andrew W. Han3, Nadir A. Mahmood3, Caitlin N. Ryan3, Yun Sok Lee1, Jeramie D. Watrous1,2, Mahendra D. Chordia4, Dongfeng Pan4, Mohit Jain1,2, Jerrold M. Olefsky1 * Affiliations: 1 Division of Endocrinology & Metabolism, Department of Medicine, University of California, San Diego, La Jolla, California, USA. 2 Department of Pharmacology, University of California, San Diego, La Jolla, California, USA. 3 Second Genome, Inc., South San Francisco, California, USA. 4 Department of Radiology and Medical Imaging, University of Virginia, Charlottesville, VA, USA. * Correspondence to: 858-534-2230, [email protected] Word Count: 4749 Figures: 6 Supplemental Figures: 11 Supplemental Tables: 5 1 Diabetes Publish Ahead of Print, published online April 22, 2019 Diabetes Page 2 of 230 ABSTRACT The composition of the gastrointestinal (GI) microbiota and associated metabolites changes dramatically with diet and the development of obesity. Although many correlations have been described, specific mechanistic links between these changes and glucose homeostasis remain to be defined. Here we show that blood and intestinal levels of the microbiota-produced N-formyl peptide, formyl-methionyl-leucyl-phenylalanine (fMLF), are elevated in high fat diet (HFD)- induced obese mice. Genetic or pharmacological inhibition of the N-formyl peptide receptor Fpr1 leads to increased insulin levels and improved glucose tolerance, dependent upon glucagon- like peptide-1 (GLP-1). Obese Fpr1-knockout (Fpr1-KO) mice also display an altered microbiome, exemplifying the dynamic relationship between host metabolism and microbiota. -

Identifying Genetic Risk Factors for Coronary Artery Angiographic Stenosis in a Genetically Diverse Population
Please do not remove this page Identifying Genetic Risk Factors for Coronary Artery Angiographic Stenosis in a Genetically Diverse Population Liu, Zhi https://scholarship.miami.edu/discovery/delivery/01UOML_INST:ResearchRepository/12355224170002976?l#13355497430002976 Liu, Z. (2016). Identifying Genetic Risk Factors for Coronary Artery Angiographic Stenosis in a Genetically Diverse Population [University of Miami]. https://scholarship.miami.edu/discovery/fulldisplay/alma991031447280502976/01UOML_INST:ResearchR epository Embargo Downloaded On 2021/09/26 20:05:11 -0400 Please do not remove this page UNIVERSITY OF MIAMI IDENTIFYING GENETIC RISK FACTORS FOR CORONARY ARTERY ANGIOGRAPHIC STENOSIS IN A GENETICALLY DIVERSE POPULATION By Zhi Liu A DISSERTATION Submitted to the Faculty of the University of Miami in partial fulfillment of the requirements for the degree of Doctor of Philosophy Coral Gables, Florida August 2016 ©2016 Zhi Liu All Rights Reserved UNIVERSITY OF MIAMI A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy IDENTIFYING GENETIC RISK FACTORS FOR CORONARY ARTERY ANGIOGRAPHIC STENOSIS IN A GENETICALLY DIVERSE POPULATION Zhi Liu Approved: ________________ _________________ Gary W. Beecham, Ph.D. Liyong Wang, Ph.D. Assistant Professor of Human Associate Professor of Human Genetics Genetics ________________ _________________ Eden R. Martin, Ph.D. Guillermo Prado, Ph.D. Professor of Human Genetics Dean of the Graduate School ________________ Tatjana Rundek, M.D., Ph.D. Professor of Neurology LIU, ZHI (Ph.D., Human Genetics and Genomics) Identifying Genetic Risk Factors for Coronary Artery (August 2016) Angiographic Stenosis in a Genetically Diverse Population Abstract of a dissertation at the University of Miami. Dissertation supervised by Professor Gary W. -

A Comprehensive Protein–Protein Interactome for Yeast PAS Kinase 1 Reveals Direct Inhibition of Respiration Through the Phosphorylation of Cbf1
M BoC | ARTICLE A comprehensive protein–protein interactome for yeast PAS kinase 1 reveals direct inhibition of respiration through the phosphorylation of Cbf1 Desiree DeMillea, Benjamin T. Bikmanb, Andrew D. Mathisc, John T. Princec, Jordan T. Mackaya, Steven W. Sowaa, Tacie D. Halla, and Julianne H. Grosea aDepartment of Microbiology and Molecular Biology, bDepartment of Physiology and Developmental Biology, and cDepartment of Chemistry, Brigham Young University, Provo, UT 84602 ABSTRACT Per-Arnt-Sim (PAS) kinase is a sensory protein kinase required for glucose ho- Monitoring Editor meostasis in yeast, mice, and humans, yet little is known about the molecular mechanisms of Charles Boone its function. Using both yeast two-hybrid and copurification approaches, we identified the University of Toronto protein–protein interactome for yeast PAS kinase 1 (Psk1), revealing 93 novel putative pro- Received: Nov 1, 2013 tein binding partners. Several of the Psk1 binding partners expand the role of PAS kinase in Revised: Apr 28, 2014 glucose homeostasis, including new pathways involved in mitochondrial metabolism. In addi- Accepted: May 12, 2014 tion, the interactome suggests novel roles for PAS kinase in cell growth (gene/protein expres- sion, replication/cell division, and protein modification and degradation), vacuole function, and stress tolerance. In vitro kinase studies using a subset of 25 of these binding partners identified Mot3, Zds1, Utr1, and Cbf1 as substrates. Further evidence is provided for the in vivo phosphorylation of Cbf1 at T211/T212 and for the subsequent inhibition of respiration. This respiratory role of PAS kinase is consistent with the reported hypermetabolism of PAS kinase–deficient mice, identifying a possible molecular mechanism and solidifying the evolu- tionary importance of PAS kinase in the regulation of glucose homeostasis. -

Intimate Relations—Mitochondria and Ageing
International Journal of Molecular Sciences Review Intimate Relations—Mitochondria and Ageing Michael Webb and Dionisia P. Sideris * Mitobridge Inc., an Astellas Company, 1030 Massachusetts Ave, Cambridge, MA 02138, USA; [email protected] * Correspondence: [email protected] Received: 29 August 2020; Accepted: 6 October 2020; Published: 14 October 2020 Abstract: Mitochondrial dysfunction is associated with ageing, but the detailed causal relationship between the two is still unclear. Wereview the major phenomenological manifestations of mitochondrial age-related dysfunction including biochemical, regulatory and energetic features. We conclude that the complexity of these processes and their inter-relationships are still not fully understood and at this point it seems unlikely that a single linear cause and effect relationship between any specific aspect of mitochondrial biology and ageing can be established in either direction. Keywords: mitochondria; ageing; energetics; ROS; gene regulation 1. Introduction The last two decades have witnessed a dramatic transformation in our view of mitochondria, their basic biology and functions. While still regarded as functioning primarily as the eukaryotic cell’s generator of energy in the form of adenosine triphosphate (ATP) and nicotinamide adenine dinucleotide (reduced form; NADH), mitochondria are now recognized as having a plethora of functions, including control of apoptosis, regulation of calcium, forming a signaling hub and the synthesis of various bioactive molecules. Their biochemical functions beyond ATP supply include biosynthesis of lipids and amino acids, formation of iron sulphur complexes and some stages of haem biosynthesis and the urea cycle. They exist as a dynamic network of organelles that under normal circumstances undergo a constant series of fission and fusion events in which structural, functional and encoding (mtDNA) elements are subject to redistribution throughout the network. -
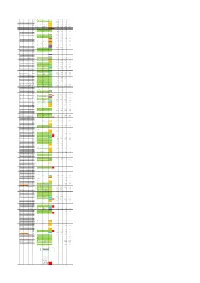
End Strand of Gene Gene Name Gene Function Starnd of Trascript
Genomic TSS strand Gene starnd of Transcipt relatively to TSS Start End Gene name coordinates relatively TSS type Control_fwd TEX_fwd Control_rev TEX_rev of gene function trascript gene group of TSS to ATG ribosomal 93 854 + rps12 + 1 protein S12 1 1 14.0638317 20.7539794 0 0 93 324 exon 1 + 111 rps12 intron 1 1 13.90756687 18.07149224 0.781323982 0.423550599 829 854 exon 2 + 496 rps12 exon2 2 24.22104343 15.24782157 0 0 30S 904 1371 + gene rps7 ribosomal + 2 protein S7 1303 -209 rps7 inter (ndhB) 2 20.47068832 9.035746118 0.625059185 0.847101199 NADH dehydrogen 1512 3535 + gene ndhB (ndh2) + ase subunit 3 2 1696 ndhB exon 1-inter 2 3.594090315 2.964854195 0.468794389 0.282367066 + 2209 ndhB exon 1-inter 2 46.09811492 4.09432246 0.468794389 0.423550599 1512 2237 exon 1 + 2756 ndhB exon 2 -inter 2 43.28534858 4.800240125 0.312529593 0.282367066 2756 3535 exon 2 + 3090 ndhB exon 2 -inter 2 17.50165719 15.95373924 0.312529593 0 + 3192 ndhB exon 2 -inter 2 140.6383167 117.6058831 2.812766334 1.694202397 - 3462 ndhB exon 2 -inter 3 1.406383167 1.129468265 1.406383167 3.67077186 4 3633 3712 + tRNA tRNA-Leu-CAA + 3610 -23 tRNA-Leu 1 77.19480938 84.85130339 0.625059185 0 + 3633 1 tRNA-Leu 1 359.5652963 649.0207016 0.781323982 0 photosyste 3954 4058 + gene psbM m II protein + 5 M 3775 -179 psbM 2 20.47068832 12.00060031 0 0.141183533 + 3954 psbM-0 2 69.22530477 28.37789015 0.156264796 0 hypothetical 4182 5073 + gene ycf66 6 protein 4182 4287 exon 1 4772 5073 exon 2 7 5202 5113 - gene ycf (ORF29) - 5299 orf29 inter 1 0 0 3.125295926 3.67077186 -

WO 2016/200987 Al 15 December 2016 (15.12.2016) P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2016/200987 Al 15 December 2016 (15.12.2016) P O P C T (51) International Patent Classification: Trudi, A.; Indigo Agriculture, Inc., 500 Rutherford Aven A01N 63/02 (2006.01) A01C 1/00 (2006.01) ue, North Building, Boston, MA 02129 (US). A01N 63/00 (2006.01) A01C 1/06 (2006.01) (74) Agents: HUBL, Susan, T. et al; Fenwick & West LLP, (21) International Application Number: Silicon Valley Center, 801 California Street, Mountain PCT/US20 16/036504 View, CA 94041 (US). (22) International Filing Date: (81) Designated States (unless otherwise indicated, for every 8 June 2016 (08.06.2016) kind of national protection available): AE, AG, AL, AM, AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, (25) Filing Language: English BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, (26) Publication Language: English DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, HN, HR, HU, ID, IL, N , IR, IS, JP, KE, KG, KN, KP, KR, (30) Priority Data: KZ, LA, LC, LK, LR, LS, LU, LY, MA, MD, ME, MG, 62/172,748 8 June 2015 (08.06.2015) US MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, OM, 62/172,750 8 June 2015 (08.06.2015) US PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, SC, 62/172,755 8 June 2015 (08.06.2015) us SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, 62/3 16,386 31 March 2016 (3 1.03.2016) us TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. -

Uncovering the Human Methyltransferasome*DS
Research © 2011 by The American Society for Biochemistry and Molecular Biology, Inc. This paper is available on line at http://www.mcponline.org Uncovering the Human Methyltransferasome*□S Tanya C. Petrossian and Steven G. Clarke‡ We present a comprehensive analysis of the human meth- core (2, 3, 5, 6, 15). The SPOUT methyltransferase superfamily yltransferasome. Primary sequences, predicted second- contains a distinctive knot structure and methylates RNA ary structures, and solved crystal structures of known substrates (16). SET domain methyltransferases catalyze the methyltransferases were analyzed by hidden Markov methylation of protein lysine residues with histones and ribo- models, Fisher-based statistical matrices, and fold recog- somal proteins as major targets (17–19). Smaller superfamilies nition prediction-based threading algorithms to create a with at least one three-dimensional structure available include model, or profile, of each methyltransferase superfamily. the precorrin-like methyltransferases (20), the radical SAM1 These profiles were used to scan the human proteome methyltransferases (21, 22), the MetH activation domain (23), database and detect novel methyltransferases. 208 pro- teins in the human genome are now identified as known or the Tyw3 protein involved in wybutosine synthesis (24), and putative methyltransferases, including 38 proteins that the homocysteine methyltransferases (25–27). Lastly, an inte- were not annotated previously. To date, 30% of these gral membrane methyltransferase family has been defined