In Vitro Reconstitution of Functional Small Ribosomal Subunit Assembly for Comprehensive Analysis of Ribosomal Elements in E. Coli
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The DEAD-Box RNA Helicase-Like Utp25 Is an SSU Processome Component
Downloaded from rnajournal.cshlp.org on September 25, 2021 - Published by Cold Spring Harbor Laboratory Press The DEAD-box RNA helicase-like Utp25 is an SSU processome component J. MICHAEL CHARETTE1,2 and SUSAN J. BASERGA1,2,3 1Department of Molecular Biophysics & Biochemistry, Yale University School of Medicine, New Haven, Connecticut 06520, USA 2Department of Therapeutic Radiology, Yale University School of Medicine, New Haven, Connecticut 06520, USA 3Department of Genetics, Yale University School of Medicine, New Haven, Connecticut 06520, USA ABSTRACT The SSU processome is a large ribonucleoprotein complex consisting of the U3 snoRNA and at least 43 proteins. A database search, initiated in an effort to discover additional SSU processome components, identified the uncharacterized, conserved and essential yeast nucleolar protein YIL091C/UTP25 as one such candidate. The C-terminal DUF1253 motif, a domain of unknown function, displays limited sequence similarity to DEAD-box RNA helicases. In the absence of the conserved DEAD-box sequence, motif Ia is the only clearly identifiable helicase element. Since the yeast homolog is nucleolar and interacts with components of the SSU processome, we examined its role in pre-rRNA processing. Genetic depletion of Utp25 resulted in slowed growth. Northern analysis of pre-rRNA revealed an 18S rRNA maturation defect at sites A0,A1, and A2. Coimmunoprecipitation confirmed association with U3 snoRNA and with Mpp10, and with components of the t-Utp/UtpA, UtpB, and U3 snoRNP subcomplexes. Mutation of the conserved motif Ia residues resulted in no discernable temperature- sensitive or cold-sensitive growth defects, implying that this motif is dispensable for Utp25 function. -

Proteotoxicity from Aberrant Ribosome Biogenesis Compromises Cell Fitness
Proteotoxicity From Aberrant Ribosome Biogenesis Compromises Cell Fitness The Harvard community has made this article openly available. Please share how this access benefits you. Your story matters Citation Tye, Blake Wells. 2019. Proteotoxicity From Aberrant Ribosome Biogenesis Compromises Cell Fitness. Doctoral dissertation, Harvard University, Graduate School of Arts & Sciences. Citable link http://nrs.harvard.edu/urn-3:HUL.InstRepos:42013104 Terms of Use This article was downloaded from Harvard University’s DASH repository, and is made available under the terms and conditions applicable to Other Posted Material, as set forth at http:// nrs.harvard.edu/urn-3:HUL.InstRepos:dash.current.terms-of- use#LAA Proteotoxicity from Aberrant Ribosome Biogenesis Compromises Cell Fitness A dissertation presented by Blake Wells Tye to The Committee on Higher Degrees in Chemical Biology in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the subject of Chemical Biology Harvard University Cambridge, Massachusetts May 2019 © 2019 Blake Wells Tye All rights reserved. Dissertation Advisor: Author: L. Stirling Churchman, PhD Blake Wells Tye Proteotoxicity from Aberrant Ribosome Biogenesis Compromises Cell Fitness Abstract Proteins are the workhorses of the cell, carrying out much of the structural and enzymatic work of life. Many proteins function as part of greater assemblies, or complexes, with other proteins and/or biomolecules. As the cell grows and divides, it must double its proteome, which requires synthesis and folding of individual proteins, assembly into complexes, and proper subcellular localization. This is repeated for millions of proteins molecules compromising thousands of potential assemblies, making up a rather tricky set of tasks for cells to carry out simultaneously. -

Domain of Escherichia Coli 16S Ribosomal RNA Using Site-Directed Photoaffinity Crosslinking
Downloaded from rnajournal.cshlp.org on October 6, 2021 - Published by Cold Spring Harbor Laboratory Press RNA (1998), 4:1455–1466+ Cambridge University Press+ Printed in the USA+ Copyright © 1998 RNA Society+ Analysis of the conformation of the 39 major domain of Escherichia coli 16S ribosomal RNA using site-directed photoaffinity crosslinking ALEXANDRE MONTPETIT,1 CATHERINE PAYANT,1 JAMES M. NOLAN,2 and LÉA BRAKIER-GINGRAS1 1Département de Biochimie, Université de Montréal, Montréal, Québec H3T 1J4, Canada 2Department of Biochemistry, Tulane University Medical Center, New Orleans, Louisiana 70112, USA ABSTRACT The 39 major domain of Escherichia coli 16S rRNA, which occupies the head of the small ribosomal subunit, is involved in several functions of the ribosome. We have used a site-specific crosslinking procedure to gain further insights into the higher-order structure of this domain. Circularly permuted RNAs were used to introduce an azi- dophenacyl group at specific positions within the 39 major domain. Crosslinks were generated in a high-ionic strength buffer that has been used for ribosome reconstitution studies and so enables the RNA to adopt a structure recognized by ribosomal proteins. The crosslinking sites were identified by primer extension and confirmed by assessing the mobility of the crosslinked RNA lariats in denaturing polyacrylamide gels. Eight crosslinks were characterized. Among them, one crosslink demonstrates that helix 28 is proximal to the top of helix 34, and two others show that the 1337 region, located in an internal loop at the junction of helices 29, 30, 41, and 42, is proximal to the center of helix 30 and to a segment connecting helix 28 to helix 29. -

1 Title 1 Discovery of a Small Molecule That Inhibits Bacterial Ribosome Biogenesis 2 3 Authors and Affiliations 4 Jonathan M. S
1 Title 2 Discovery of a small molecule that inhibits bacterial ribosome biogenesis 3 4 Authors and Affiliations 5 Jonathan M. Stokes1, Joseph H. Davis2, Chand S. Mangat1, James R. Williamson2, and Eric D. Brown1* 6 7 1Michael G. DeGroote Institute for Infectious Disease Research, Department of Biochemistry and 8 Biomedical Sciences, McMaster University, Hamilton, Ontario, Canada, L8N 3Z5 9 2Department of Integrative Structural and Computational Biology, Department of Chemistry and The Skaggs 10 Institute for Chemical Biology, The Scripps Research Institute, La Jolla, CA 92037, USA 11 12 Contact 13 *Correspondence: [email protected] 14 15 Competing Interests Statement 16 All authors declare no competing interests. 17 18 Impact Statement 19 We report the discovery of a small molecule inhibitor of bacterial ribosome biogenesis. The first probe of its 20 kind, this compound revealed an unexplored role for IF2 in ribosome assembly. 21 22 Major Subject Areas 23 Biochemistry; Microbiology and infectious disease 24 25 Keywords 26 Cold sensitivity; ribosome biogenesis; lamotrigine; translation initiation factor IF2 27 28 1 29 Abstract 30 While small molecule inhibitors of the bacterial ribosome have been instrumental in understanding protein 31 translation, no such probes exist to study ribosome biogenesis. We screened a diverse chemical collection 32 that included previously approved drugs for compounds that induced cold sensitive growth inhibition in the 33 model bacterium Escherichia coli. Among the most cold sensitive was lamotrigine, an anticonvulsant drug. 34 Lamotrigine treatment resulted in the rapid accumulation of immature 30S and 50S ribosomal subunits at 35 15°C. Importantly, this was not the result of translation inhibition, as lamotrigine was incapable of perturbing 36 protein synthesis in vivo or in vitro. -

Nucleolin and Its Role in Ribosomal Biogenesis
NUCLEOLIN: A NUCLEOLAR RNA-BINDING PROTEIN INVOLVED IN RIBOSOME BIOGENESIS Inaugural-Dissertation zur Erlangung des Doktorgrades der Mathematisch-Naturwissenschaftlichen Fakultät der Heinrich-Heine-Universität Düsseldorf vorgelegt von Julia Fremerey aus Hamburg Düsseldorf, April 2016 2 Gedruckt mit der Genehmigung der Mathematisch-Naturwissenschaftlichen Fakultät der Heinrich-Heine-Universität Düsseldorf Referent: Prof. Dr. A. Borkhardt Korreferent: Prof. Dr. H. Schwender Tag der mündlichen Prüfung: 20.07.2016 3 Die vorgelegte Arbeit wurde von Juli 2012 bis März 2016 in der Klinik für Kinder- Onkologie, -Hämatologie und Klinische Immunologie des Universitätsklinikums Düsseldorf unter Anleitung von Prof. Dr. A. Borkhardt und in Kooperation mit dem ‚Laboratory of RNA Molecular Biology‘ an der Rockefeller Universität unter Anleitung von Prof. Dr. T. Tuschl angefertigt. 4 Dedicated to my family TABLE OF CONTENTS 5 TABLE OF CONTENTS TABLE OF CONTENTS ............................................................................................... 5 LIST OF FIGURES ......................................................................................................10 LIST OF TABLES .......................................................................................................12 ABBREVIATION .........................................................................................................13 ABSTRACT ................................................................................................................19 ZUSAMMENFASSUNG -

The Microbiota-Produced N-Formyl Peptide Fmlf Promotes Obesity-Induced Glucose
Page 1 of 230 Diabetes Title: The microbiota-produced N-formyl peptide fMLF promotes obesity-induced glucose intolerance Joshua Wollam1, Matthew Riopel1, Yong-Jiang Xu1,2, Andrew M. F. Johnson1, Jachelle M. Ofrecio1, Wei Ying1, Dalila El Ouarrat1, Luisa S. Chan3, Andrew W. Han3, Nadir A. Mahmood3, Caitlin N. Ryan3, Yun Sok Lee1, Jeramie D. Watrous1,2, Mahendra D. Chordia4, Dongfeng Pan4, Mohit Jain1,2, Jerrold M. Olefsky1 * Affiliations: 1 Division of Endocrinology & Metabolism, Department of Medicine, University of California, San Diego, La Jolla, California, USA. 2 Department of Pharmacology, University of California, San Diego, La Jolla, California, USA. 3 Second Genome, Inc., South San Francisco, California, USA. 4 Department of Radiology and Medical Imaging, University of Virginia, Charlottesville, VA, USA. * Correspondence to: 858-534-2230, [email protected] Word Count: 4749 Figures: 6 Supplemental Figures: 11 Supplemental Tables: 5 1 Diabetes Publish Ahead of Print, published online April 22, 2019 Diabetes Page 2 of 230 ABSTRACT The composition of the gastrointestinal (GI) microbiota and associated metabolites changes dramatically with diet and the development of obesity. Although many correlations have been described, specific mechanistic links between these changes and glucose homeostasis remain to be defined. Here we show that blood and intestinal levels of the microbiota-produced N-formyl peptide, formyl-methionyl-leucyl-phenylalanine (fMLF), are elevated in high fat diet (HFD)- induced obese mice. Genetic or pharmacological inhibition of the N-formyl peptide receptor Fpr1 leads to increased insulin levels and improved glucose tolerance, dependent upon glucagon- like peptide-1 (GLP-1). Obese Fpr1-knockout (Fpr1-KO) mice also display an altered microbiome, exemplifying the dynamic relationship between host metabolism and microbiota. -

Intimate Relations—Mitochondria and Ageing
International Journal of Molecular Sciences Review Intimate Relations—Mitochondria and Ageing Michael Webb and Dionisia P. Sideris * Mitobridge Inc., an Astellas Company, 1030 Massachusetts Ave, Cambridge, MA 02138, USA; [email protected] * Correspondence: [email protected] Received: 29 August 2020; Accepted: 6 October 2020; Published: 14 October 2020 Abstract: Mitochondrial dysfunction is associated with ageing, but the detailed causal relationship between the two is still unclear. Wereview the major phenomenological manifestations of mitochondrial age-related dysfunction including biochemical, regulatory and energetic features. We conclude that the complexity of these processes and their inter-relationships are still not fully understood and at this point it seems unlikely that a single linear cause and effect relationship between any specific aspect of mitochondrial biology and ageing can be established in either direction. Keywords: mitochondria; ageing; energetics; ROS; gene regulation 1. Introduction The last two decades have witnessed a dramatic transformation in our view of mitochondria, their basic biology and functions. While still regarded as functioning primarily as the eukaryotic cell’s generator of energy in the form of adenosine triphosphate (ATP) and nicotinamide adenine dinucleotide (reduced form; NADH), mitochondria are now recognized as having a plethora of functions, including control of apoptosis, regulation of calcium, forming a signaling hub and the synthesis of various bioactive molecules. Their biochemical functions beyond ATP supply include biosynthesis of lipids and amino acids, formation of iron sulphur complexes and some stages of haem biosynthesis and the urea cycle. They exist as a dynamic network of organelles that under normal circumstances undergo a constant series of fission and fusion events in which structural, functional and encoding (mtDNA) elements are subject to redistribution throughout the network. -
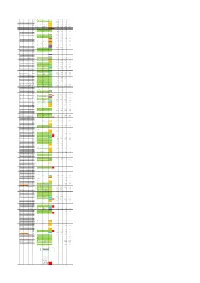
End Strand of Gene Gene Name Gene Function Starnd of Trascript
Genomic TSS strand Gene starnd of Transcipt relatively to TSS Start End Gene name coordinates relatively TSS type Control_fwd TEX_fwd Control_rev TEX_rev of gene function trascript gene group of TSS to ATG ribosomal 93 854 + rps12 + 1 protein S12 1 1 14.0638317 20.7539794 0 0 93 324 exon 1 + 111 rps12 intron 1 1 13.90756687 18.07149224 0.781323982 0.423550599 829 854 exon 2 + 496 rps12 exon2 2 24.22104343 15.24782157 0 0 30S 904 1371 + gene rps7 ribosomal + 2 protein S7 1303 -209 rps7 inter (ndhB) 2 20.47068832 9.035746118 0.625059185 0.847101199 NADH dehydrogen 1512 3535 + gene ndhB (ndh2) + ase subunit 3 2 1696 ndhB exon 1-inter 2 3.594090315 2.964854195 0.468794389 0.282367066 + 2209 ndhB exon 1-inter 2 46.09811492 4.09432246 0.468794389 0.423550599 1512 2237 exon 1 + 2756 ndhB exon 2 -inter 2 43.28534858 4.800240125 0.312529593 0.282367066 2756 3535 exon 2 + 3090 ndhB exon 2 -inter 2 17.50165719 15.95373924 0.312529593 0 + 3192 ndhB exon 2 -inter 2 140.6383167 117.6058831 2.812766334 1.694202397 - 3462 ndhB exon 2 -inter 3 1.406383167 1.129468265 1.406383167 3.67077186 4 3633 3712 + tRNA tRNA-Leu-CAA + 3610 -23 tRNA-Leu 1 77.19480938 84.85130339 0.625059185 0 + 3633 1 tRNA-Leu 1 359.5652963 649.0207016 0.781323982 0 photosyste 3954 4058 + gene psbM m II protein + 5 M 3775 -179 psbM 2 20.47068832 12.00060031 0 0.141183533 + 3954 psbM-0 2 69.22530477 28.37789015 0.156264796 0 hypothetical 4182 5073 + gene ycf66 6 protein 4182 4287 exon 1 4772 5073 exon 2 7 5202 5113 - gene ycf (ORF29) - 5299 orf29 inter 1 0 0 3.125295926 3.67077186 -

WO 2016/200987 Al 15 December 2016 (15.12.2016) P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2016/200987 Al 15 December 2016 (15.12.2016) P O P C T (51) International Patent Classification: Trudi, A.; Indigo Agriculture, Inc., 500 Rutherford Aven A01N 63/02 (2006.01) A01C 1/00 (2006.01) ue, North Building, Boston, MA 02129 (US). A01N 63/00 (2006.01) A01C 1/06 (2006.01) (74) Agents: HUBL, Susan, T. et al; Fenwick & West LLP, (21) International Application Number: Silicon Valley Center, 801 California Street, Mountain PCT/US20 16/036504 View, CA 94041 (US). (22) International Filing Date: (81) Designated States (unless otherwise indicated, for every 8 June 2016 (08.06.2016) kind of national protection available): AE, AG, AL, AM, AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, (25) Filing Language: English BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, (26) Publication Language: English DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, HN, HR, HU, ID, IL, N , IR, IS, JP, KE, KG, KN, KP, KR, (30) Priority Data: KZ, LA, LC, LK, LR, LS, LU, LY, MA, MD, ME, MG, 62/172,748 8 June 2015 (08.06.2015) US MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, OM, 62/172,750 8 June 2015 (08.06.2015) US PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, SC, 62/172,755 8 June 2015 (08.06.2015) us SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, 62/3 16,386 31 March 2016 (3 1.03.2016) us TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. -

Transcriptomic Regulation of Alternative Phenotypic Trajectories in Embryos of the Annual Killifish Austrofundulus Limnaeus
Portland State University PDXScholar Dissertations and Theses Dissertations and Theses Fall 11-30-2017 Transcriptomic Regulation of Alternative Phenotypic Trajectories in Embryos of the Annual Killifish Austrofundulus limnaeus Amie L. Romney Portland State University Follow this and additional works at: https://pdxscholar.library.pdx.edu/open_access_etds Part of the Biology Commons, and the Genetics and Genomics Commons Let us know how access to this document benefits ou.y Recommended Citation Romney, Amie L., "Transcriptomic Regulation of Alternative Phenotypic Trajectories in Embryos of the Annual Killifish Austrofundulus limnaeus" (2017). Dissertations and Theses. Paper 4033. https://doi.org/10.15760/etd.5917 This Dissertation is brought to you for free and open access. It has been accepted for inclusion in Dissertations and Theses by an authorized administrator of PDXScholar. Please contact us if we can make this document more accessible: [email protected]. Transcriptomic Regulation of Alternative Phenotypic Trajectories in embryos of the Annual Killifish Austrofundulus limnaeus by Amie Lynn Thomas Romney A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in Biology Dissertation Committee Jason Podrabsky, Chair Suzanne Estes Bradley Buckley Todd Rosenstiel Dirk Iwata-Reuyl Portland State University 2017 © 2017 Amie Lynn Thomas Romney ABSTRACT The Annual Killifish, Austrofundulus limnaeus, survives the seasonal drying of their pond habitat in the form of embryos entering diapause midway through development. The diapause trajectory is one of two developmental phenotypes. Alternatively, individuals can “escape” entry into diapause and develop continuously until hatching. The alternative phenotypes of A. limnaeus are a form of developmental plasticity that provides this species with a physiological adaption for surviving stressful environments. -

Open FINAL GRAD SCHOOL.Pdf
The Pennsylvania State University The Graduate School Eberly College of Science PHOSPHOPROTEOMIC ANALYSIS OF RIBOSOMAL PROTEINS: IMPLICATIONS IN TRANSLATION AND APOPTOSIS A Dissertation in Biochemistry, Microbiology, and Molecular Biology by Jennifer Lynn Miller © 2009 Jennifer Lynn Miller Submitted in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy May 2009 The dissertation of Jennifer Lynn Miller was reviewed and approved* by the following: Emine C. Koc Assistant Professor Biochemistry and Molecular Biology Dissertation Advisor Chair of Committee Robert A. Schlegel Professor of Biochemistry and Molecular Biology Wendy Hanna-Rose Assistant Professor Biochemistry and Molecular Biology Ming Tien Professor of Biochemistry Erin D. Sheets Assistant Professor of Chemistry Richard J. Frisque Professor of Molecular Virology Head of the Department of Biochemistry and Molecular Biology *Signatures are on file in the Graduate School. ABSTRACT Mammalian mitochondrial ribosomes synthesize thirteen proteins that are essential for oxidative phosphorylation. Besides having a major role in ATP synthesis, mitochondria also contribute to biochemical processes coordinating apoptosis, mitochondrial diseases, and aging in eukaryotic cells. This unique class of ribosomes is protein-rich and distinct from cytoplasmic ribosomes. However, mitochondrial ribosomes (55S) share a significant homology to bacterial ribosomes (70S), particularly in size, the general mechanism of translation, and ribosomal protein content. Due to the overall resemblance between the two systems and the earlier reports of post-translational modifications, we investigated how phosphorylation of ribosomal proteins from bacteria and mitochondria regulates translation and other acquired roles. Identification of twenty- four phosphorylated 70S and 55S ribosomal proteins as well as the potential endogenous kinase was achieved using 2D-gel electrophoresis and tandem mass spectrometry. -

Translation Factors and Ribosomal Proteins Control Tumor Onset and Progression: How?
Oncogene (2014) 33, 2145–2156 & 2014 Macmillan Publishers Limited All rights reserved 0950-9232/14 www.nature.com/onc REVIEW Translation factors and ribosomal proteins control tumor onset and progression: how? F Loreni1, M Mancino2,3 and S Biffo2,3 Gene expression is shaped by translational control. The modalities and the extent by which translation factors modify gene expression have revealed therapeutic scenarios. For instance, eukaryotic initiation factor (eIF)4E activity is controlled by the signaling cascade of growth factors, and drives tumorigenesis by favoring the translation of specific mRNAs. Highly specific drugs target the activity of eIF4E. Indeed, the antitumor action of mTOR complex 1 (mTORc1) blockers like rapamycin relies on their capability to inhibit eIF4E assembly into functional eIF4F complexes. eIF4E biology, from its inception to recent pharmacological targeting, is proof-of-principle that translational control is druggable. The case for eIF4E is not isolated. The translational machinery is involved in the biology of cancer through many other mechanisms. First, untranslated sequences on mRNAs as well as noncoding RNAs regulate the translational efficiency of mRNAs that are central for tumor progression. Second, other initiation factors like eIF6 show a tumorigenic potential by acting downstream of oncogenic pathways. Third, genetic alterations in components of the translational apparatus underlie an entire class of inherited syndromes known as ‘ribosomopathies’ that are associated with increased cancer risk. Taken together, data suggest that in spite of their evolutionary conservation and ubiquitous nature, variations in the activity and levels of ribosomal proteins and translation factors generate highly specific effects. Beside, as the structures and biochemical activities of several noncoding RNAs and initiation factors are known, these factors may be amenable to rational pharmacological targeting.