Signature Redacted
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Genomic Analysis of Bone Marrow Failure and Myelodysplastic Syndromes Reveals Phenotypic and Diagnostic Complexity
Bone Marrow Failure SUPPLEMENTARY APPENDIX Genomic analysis of bone marrow failure and myelodysplastic syndromes reveals phenotypic and diagnostic complexity Michael Y. Zhang,1 Siobán B. Keel,2 Tom Walsh,3 Ming K. Lee,3 Suleyman Gulsuner,3 Amanda C. Watts,3 Colin C. Pritchard,4 Stephen J. Salipante,4 Michael R. Jeng,5 Inga Hofmann,6 David A. Williams,6,7 Mark D. Fleming,8 Janis L. Abkowitz,2 Mary-Claire King,3 and Akiko Shimamura1,9,10 M.Y.Z. and S.B.K. contributed equally to this work. 1Clinical Research Division, Fred Hutchinson Cancer Research Center, Seattle, WA; 2Department of Medicine, Division of Hematology, University of Washington, Seattle, WA; 3Department of Medicine and Department of Genome Sciences, University of Washington, Seattle, WA; 4Department of Laboratory Medicine, University of Washington, Seattle, WA; 5Department of Pediatrics, Stanford Uni- versity School of Medicine, Stanford, CA; 6Division of Hematology/Oncology, Boston Children’s Hospital, Dana Farber Cancer Insti- tute, and Harvard Medical School, Boston, MA; 7Harvard Stem Cell Institute, Boston, MA; 8Department of Pathology, Boston Children’s Hospital, MA; 9Department of Pediatric Hematology/Oncology, Seattle Children’s Hospital, WA; 10Department of Pedi- atrics, University of Washington, Seattle, WA, USA ©2014 Ferrata Storti Foundation. This is an open-access paper. doi:10.3324/haematol.2014.113456 Manuscript received on July 22, 2014. Manuscript accepted on September 15, 2014. Correspondence: [email protected] Supplementary Methods Genomics. Libraries were prepared in 96-well format with a Bravo liquid handling robot (Agilent Technologies). One to two micrograms of genomic DNA were sheared to a peak size of 150 bp using a Covaris E series instrument. -

Francois Jacob Memorial
RETROSPECTIVE RETROSPECTIVE Francois Jacob memorial Arthur B. Pardee1 Department of Adult Oncology, Dana-Farber Institute, Boston, MA 02115 Dr. Francois Jacob is one of a handful of the DNA would integrate into the bacterial chro- 20th century’smostdistinguishedlifescien- mosome and remain dormant or, at other tists. His research with Dr. Jacques Monod, times, would kill the cell. like that of Watson and Crick, provided the Jacob’s next major contribution, in collab- foundations for understanding mechanisms oration with Dr. Jacques Monod, was to in- of genetic regulation of life processes such vestigate how a gene is regulated. Remark- as cell differentiation and defects in diseases. ably, native E. coli synthesize β-galactosidase Jacob joined the College de France in 1964 only when lactose is available. Some mutated and shared the Nobel Prize in Physiology bacteria can make the enzyme in the absence or Medicine 1965 with Jacques Monod of inducer. Monod’s initial idea was that and Andre Lwoff. He was elected to the these constitutive bacteria activate the gene National Academy of Sciences (NAS) USA by synthesizing an intracellular lactose-like in 1969. inducer molecule. Jacob was born in 1920 in a French Jewish To investigate this model, interrupted family; his grandfather was a four-star gen- mating was applied to bring the β-galactosi- eral. He began to study medicine before dase gene of a donor bacterium into a consti- World War II, in which he served as a mil- tutive receptor. According to the induction itary officer in the Free French Army and was model, the mated cell should produce en- badly wounded in an air raid. -
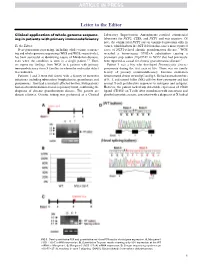
Clinical Application of Whole-Genome Sequencing in Patients with Primary
Letter to the Editor Clinical application of whole-genome sequenc- Laboratory Improvement Amendments–certified commercial ing in patients with primary immunodeficiency laboratory for NCF2, CYBA, and NCF1 and was negative. Of note, the commercial NCF1 screen examined mutations only in To the Editor: exon 2, which harbors the 2GT deletion that causes most reported Next-generation sequencing, including whole-exome sequenc- cases of NCF1-related chronic granulomatous disease.4 WGS ing and whole-genome sequencing (WES and WGS, respectively), revealed a homozygous 579G>A substitution causing a has been successful at identifying causes of Mendelian diseases, premature stop codon (Trp193X) in NCF1 that had previously even when the condition is seen in a single patient.1-3 Here, been reported as causal for chronic granulomatous disease.5 we report our findings from WGS in 6 patients with primary Patient 3 was a boy who developed Pneumocystis jiroveci immunodeficiency from 5 families in whom the molecular defect pneumonia during the first year of life. There was no family was unknown. history of primary immunodeficiency. Immune evaluation Patients 1 and 2 were full sisters with a history of recurrent demonstrated absent serum IgG and IgA. He had normal numbers infections, including tuberculous lymphadenitis, granulomas, and of B, T, and natural killer (NK) cells by flow cytometry and had pneumonias. They had a similarly affected brother. Both patients normal T-cell proliferative responses to mitogens and antigens. had an absent rhodamine-based respiratory burst, confirming the However, the patient lacked any detectable expression of CD40 diagnosis of chronic granulomatous disease. The parents are ligand (CD154) on T cells after stimulation with ionomycin and distant relatives. -

Quiet Debut'' of the Double Helix: a Bibliometric and Methodological
Journal of the History of Biology Ó Springer 2009 DOI 10.1007/s10739-009-9183-2 Revisiting the ‘‘Quiet Debut’’ of the Double Helix: A Bibliometric and Methodological note on the ‘‘Impact’’ of Scientific Publications YVES GINGRAS De´partement d’histoire Universite´ du Que´bec a` Montre´al C.P. 8888, Suc. Centre-Ville Montreal, QC H3C-3P8 Canada E-mail: [email protected] Abstract. The object of this paper is two-fold: first, to show that contrary to what seem to have become a widely accepted view among historians of biology, the famous 1953 first Nature paper of Watson and Crick on the structure of DNA was widely cited – as compared to the average paper of the time – on a continuous basis from the very year of its publication and over the period 1953–1970 and that the citations came from a wide array of scientific journals. A systematic analysis of the bibliometric data thus shows that Watson’s and Crick’s paper did in fact have immediate and long term impact if we define ‘‘impact’’ in terms of comparative citations with other papers of the time. In this precise sense it did not fall into ‘‘relative oblivion’’ in the scientific community. The second aim of this paper is to show, using the case of the reception of the Watson–Crick and Jacob–Monod papers as concrete examples, how large scale bibliometric data can be used in a sophisticated manner to provide information about the dynamic of the scientific field as a whole instead of limiting the analysis to a few major actors and generalizing the result to the whole community without further ado. -

In Silico Tools for Splicing Defect Prediction: a Survey from the Viewpoint of End Users
© American College of Medical Genetics and Genomics REVIEW In silico tools for splicing defect prediction: a survey from the viewpoint of end users Xueqiu Jian, MPH1, Eric Boerwinkle, PhD1,2 and Xiaoming Liu, PhD1 RNA splicing is the process during which introns are excised and informaticians in relevant areas who are working on huge data sets exons are spliced. The precise recognition of splicing signals is critical may also benefit from this review. Specifically, we focus on those tools to this process, and mutations affecting splicing comprise a consider- whose primary goal is to predict the impact of mutations within the able proportion of genetic disease etiology. Analysis of RNA samples 5′ and 3′ splicing consensus regions: the algorithms used by different from the patient is the most straightforward and reliable method to tools as well as their major advantages and disadvantages are briefly detect splicing defects. However, currently, the technical limitation introduced; the formats of their input and output are summarized; prohibits its use in routine clinical practice. In silico tools that predict and the interpretation, evaluation, and prospection are also discussed. potential consequences of splicing mutations may be useful in daily Genet Med advance online publication 21 November 2013 diagnostic activities. In this review, we provide medical geneticists with some basic insights into some of the most popular in silico tools Key Words: bioinformatics; end user; in silico prediction tool; for splicing defect prediction, from the viewpoint of end users. Bio- medical genetics; splicing consensus region; splicing mutation INTRODUCTION TO PRE-mRNA SPLICING AND small nuclear ribonucleoproteins and more than 150 proteins, MUTATIONS AFFECTING SPLICING serine/arginine-rich (SR) proteins, heterogeneous nuclear ribo- Sixty years ago, the milestone discovery of the double-helix nucleoproteins, and the regulatory complex (Figure 1). -

MCDB 5220 Methods and Logics April 21 2015 Marcelo Bassalo
Cracking the Genetic Code MCDB 5220 Methods and Logics April 21 2015 Marcelo Bassalo The DNA Saga… so far Important contributions for cracking the genetic code: • The “transforming principle” (1928) Frederick Griffith The DNA Saga… so far Important contributions for cracking the genetic code: • The “transforming principle” (1928) • The nature of the transforming principle: DNA (1944 - 1952) Oswald Avery Alfred Hershey Martha Chase The DNA Saga… so far Important contributions for cracking the genetic code: • The “transforming principle” (1928) • The nature of the transforming principle: DNA (1944 - 1952) • X-ray diffraction and the structure of proteins (1951) Linus Carl Pauling The DNA Saga… so far Important contributions for cracking the genetic code: • The “transforming principle” (1928) • The nature of the transforming principle: DNA (1944 - 1952) • X-ray diffraction and the structure of proteins (1951) • The structure of DNA (1953) James Watson and Francis Crick The DNA Saga… so far Important contributions for cracking the genetic code: • The “transforming principle” (1928) • The nature of the transforming principle: DNA (1944 - 1952) • X-ray diffraction and the structure of proteins (1951) • The structure of DNA (1953) How is DNA (4 nucleotides) the genetic material while proteins (20 amino acids) are the building blocks? ? DNA Protein ? The Coding Craze ? DNA Protein What was already known? • DNA resides inside the nucleus - DNA is not the carrier • Protein synthesis occur in the cytoplasm through ribosomes {• Only RNA is associated with ribosomes (no DNA) - rRNA is not the carrier { • Ribosomal RNA (rRNA) was a homogeneous population The “messenger RNA” hypothesis François Jacob Jacques Monod The Coding Craze ? DNA RNA Protein RNA Tie Club Table from Wikipedia The Coding Craze Who won the race Marshall Nirenberg J. -

The Eighth Day of Creation”: Looking Back Across 40 Years to the Birth of Molecular Biology and the Roots of Modern Cell Biology
“The Eighth Day of Creation”: looking back across 40 years to the birth of molecular biology and the roots of modern cell biology Mark Peifer1 1 Department of Biology and Curriculum in Genetics and Molecular Biology, University of North Carolina at Chapel Hill, CB#3280, Chapel Hill, NC 27599-3280, USA * To whom correspondence should be addressed Email: [email protected] Phone: (919) 962-2272 1 Forty years ago, Horace Judson’s “The Eight Day of Creation” was published, a book vividly recounting the foundations of modern biology, the molecular biology revolution. This book inspired many in my generation. The anniversary provides a chance for a new generation to take a look back, to see how science has changed and hasn’t changed. Many central players in the book, including Sydney Brenner, Seymour Benzer and Francois Jacob, would go on to be among the founders of modern cell, developmental, and neurobiology. These players come alive via their own words, as complex individuals, both heroes and anti-heroes. The technologies and experimental approaches they pioneered, ranging from cell fractionation to immunoprecipitation to structural biology, and the multidisciplinary approaches they took continue to power and inspire our work today. In the process, Judson brings out of the shadows the central roles played by women in many of the era’s discoveries. He provides us with a vision of how science and scientists have changed, of how many things about our endeavor never change, and how some new ideas are perhaps not as new as we’d like to think. 2 In 1979 Horace Judson completed a ten-year project about cell and molecular biology’s foundations, unveiling “The Eighth Day of Creation”, a book I view as one of the most masterful evocations of a scientific revolution (Judson, 1979). -

Genetic Features of Myelodysplastic Syndrome and Aplastic Anemia in Pediatric and Young Adult Patients
Bone Marrow Failure SUPPLEMENTARY APPENDIX Genetic features of myelodysplastic syndrome and aplastic anemia in pediatric and young adult patients Siobán B. Keel, 1* Angela Scott, 2,3,4 * Marilyn Sanchez-Bonilla, 5 Phoenix A. Ho, 2,3,4 Suleyman Gulsuner, 6 Colin C. Pritchard, 7 Janis L. Abkowitz, 1 Mary-Claire King, 6 Tom Walsh, 6** and Akiko Shimamura 5** 1Department of Medicine, Division of Hematology, University of Washington, Seattle, WA; 2Clinical Research Division, Fred Hutchinson Can - cer Research Center, Seattle, WA; 3Department of Pediatric Hematology/Oncology, Seattle Children’s Hospital, WA; 4Department of Pedi - atrics, University of Washington, Seattle, WA; 5Boston Children’s Hospital, Dana Farber Cancer Institute, and Harvard Medical School, MA; 6Department of Medicine and Department of Genome Sciences, University of Washington, Seattle, WA; and 7Department of Laboratory Medicine, University of Washington, Seattle, WA, USA *SBK and ASc contributed equally to this work **TW and ASh are co-senior authors ©2016 Ferrata Storti Foundation. This is an open-access paper. doi:10.3324/haematol. 2016.149476 Received: May 16, 2016. Accepted: July 13, 2016. Pre-published: July 14, 2016. Correspondence: [email protected] or [email protected] Supplementary materials Supplementary methods Retrospective chart review Patient data were collected from medical records by two investigators blinded to the results of genetic testing. The following information was collected: date of birth, transplant, death, and last follow-up, -

Spectrum of Splicing Errors Caused by CHRNE Mutations Affecting Introns and Intron/Exon Boundaries K Ohno, a Tsujino, X-M Shen, M Milone, a G Engel
1of5 ONLINE MUTATION REPORT J Med Genet: first published as 10.1136/jmg.2004.026682 on 1 August 2005. Downloaded from Spectrum of splicing errors caused by CHRNE mutations affecting introns and intron/exon boundaries K Ohno, A Tsujino, X-M Shen, M Milone, A G Engel ............................................................................................................................... J Med Genet 2005;42:e53 (http://www.jmedgenet.com/cgi/content/full/42/8/e53). doi: 10.1136/jmg.2004.026682 Patients Background: Mutations in CHRNE, the gene encoding the Patients 1–5 (respectively a 59 year old woman, a 23 year old muscle nicotinic acetylcholine receptor e subunit, cause man, a 2.5 year old girl, a 6 year old boy, and a 44 year old congenital myasthenic syndromes. Only three of the eight man) have moderate to severe myasthenic symptoms that intronic splice site mutations of CHRNE reported to date have have been present since birth or infancy, decremental EMG had their splicing consequences characterised. responses, and no AChR antibodies. All respond partially to Methods: We analysed four previously reported and five pyridostigmine. Patient 4 underwent an intercostal muscle novel splicing mutations in CHRNE by introducing the entire biopsy for diagnosis, which showed severe endplate AChR normal and mutant genomic CHRNEs into COS cells. deficiency (6% of normal) and compensatory expression of Results and conclusions: We found that short introns (82– the fetal c-AChR at the endplate. 109 nucleotides) favour intron retention, whereas medium to long introns (306–1210 nucleotides) flanking either or both Construction of CHRNE clones for splicing analysis sides of an exon favour exon skipping. Two mutations are of To examine the consequences of the identified splice site particular interest. -

In 1953 in England James Watson and Francis Crick Discovered the Structure of DNA in the Now-Famous Scientific Narrative Known As the “Race Towards the Double Helix”
THE NARRATIVES OF SCIENCE: LITERARY THEORY AND DISCOVERY IN MOLECULAR BIOLOGY PRIYA VENKATESAN In 1953 in England James Watson and Francis Crick discovered the structure of DNA in the now-famous scientific narrative known as the “race towards the double helix”. Meanwhile in France, Roland Barthes published his first book, Writing Degree Zero, on literary theory, which became the intellectual precursor for the new human sciences that were developing based on Saussurean linguistics. The discovery by Watson and Crick of the double helix marked a definitive turning point in the development of the life sciences, paving the way for the articulation of the genetic code and the emergence of molecular biology. The publication by Barthes was no less significant, since it served as an exemplar for elucidating how literary narratives are structured and for formulating how textual material is constructed. As Françoise Dosse notes, Writing Degree Zero “received unanimous acclaim and quickly became a symptom of new literary demands, a break with tradition”.1 Both the work of Roland Barthes and Watson and Crick served as paradigms in their respective fields. Semiotics, the field of textual analysis as developed by Barthes in Writing Degree Zero, offered a new direction in the structuring of narrative whereby each distinct unit in a story formed a “code” or “isotopy” that categorizes the formal elements of the story. The historical concurrence of the discovery of the double helix and the publication of Writing Degree Zero may be mere coincidence, but this essay is an exploration of the intellectual influence that both events may have had on each other, since both the discovery of the double helix and Barthes’ publication gave expression to the new forms of knowledge 1 Françoise Dosse, History of Structuralism: The Rising Sign, 1945-1966, trans. -

Background Splicing and Genetic Disease
Background splicing and genetic disease Diana Alexieva Imperial College London Yi Long Imperial College London Rupa Sarkar Imperial College London Hansraj Dhayan Imperial College London Emmanuel Bruet Imperial College London Robert Winston Imperial College London Igor Vorechovsky University of Southampton Leandro Castellano University of Sussex Nick Dibb ( [email protected] ) Imperial College London Research Article Keywords: background splicing, splice site mutations, cryptic splice sites, exon skipping, pseudoexons, recursive splicing, spliceosomal mutations, splicing therapy, BRCA1, BRCA1, DMD Posted Date: October 15th, 2020 DOI: https://doi.org/10.21203/rs.3.rs-92665/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 1/23 Abstract We report that low level background splicing by normal genes can be used to predict the likely effect of splicing mutations upon cryptic splice site activation and exon skipping, with emphasis on the DBASS databases, BRCA1, BRCA2 and DMD. In addition we show that background RNA splice sites are also involved in pseudoexon formation, recursive splicing and aberrant splicing in cancer. We discuss how background splicing information might inform splicing therapy. Introduction We previously established that cryptic splices sites (css) are already active, albeit at very low levels, in normal genes. We did this by using EST data to identify rare splice sites and then compared their positions to known css that are activated in human disease (1). However, this approach was limited to a minority of genes for which there was sucient EST sequence data. Since that time a large amount of RNA-sequencing data has been deposited, which we reasoned would strongly increase the power of css prediction. -

Cover June 2011
z NOBEL LAUREATES IN Qui DNA RESEARCH n u SANGRAM KESHARI LENKA & CHINMOYEE MAHARANA F 1. Who got the Nobel Prize in Physiology or Medicine 1933) for discovering the famous concept that says chromosomes carry genes? a. Gregor Johann Mendel b. Thomas Hunt Morgan c. Aristotle d. Charles Darwin 5. Name the Nobel laureate (1959) for his discovery of the mechanisms in the biological 2. The concept of Mutations synthesis of ribonucleic acid and are changes in genetic deoxyribonucleic acid? information” awarded him a. Arthur Kornberg b. Har Gobind Khorana the Nobel Prize in 1946: c. Roger D. Kornberg d. James D. Watson a. Hermann Muller b. M.F. Perutz c. James D. Watson 6. Discovery of the DNA double helix fetched them d. Har Gobind Khorana the Nobel Prize in Physiology or Medicine (1962). a. Francis Crick, James Watson, Rosalind Elsie Franklin b. Francis Crick, James Watson and Maurice Willkins c. James Watson, Maurice Willkins, Rosalind Elsie Franklin 3. Identify the discoverer and d. Maurice Willkins, Rosalind Elsie Franklin and Francis Crick Nobel laureate of 1958 who found DNA in bacteria and viruses. a. Louis Pasteur b. Alexander Fleming c. Joshua Lederberg d. Roger D. Kornberg 4. A direct link between genes and enzymatic reactions, known as the famous “one gene, one enzyme” hypothesis, was put forth by these 7. They developed the theory of genetic regulatory scientists who shared the Nobel Prize in mechanisms, showing how, on a molecular level, Physiology or Medicine, 1958. certain genes are activated and suppressed. Name a. George Wells Beadle and Edward Lawrie Tatum these famous Nobel laureates of 1965.