The Etiology and Molecular Genetics of Human Pigmentation Disorders
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Metastasizing Melanoma Formation Caused by Expression of Activated N-Rasq61k on an Ink4a-Deficient Background
Research Article Metastasizing Melanoma Formation Caused by Expression of Activated N-RasQ61K on an INK4a-Deficient Background Julien Ackermann,1 Manon Frutschi,1 Kostas Kaloulis,1 Thomas McKee,2 Andreas Trumpp,1 and Friedrich Beermann1 1ISREC, Swiss Institute for Experimental Cancer Research, National Center of Competence in Research Molecular Oncology, Epalinges, Switzerland and 2Institute of Pathology, University of Lausanne, Lausanne, Switzerland Abstract the most common factors predisposing to melanoma formation in humans is the INK4a/ARF locus (4), which is inactivated with high In human cutaneous malignant melanoma, a predominance of frequency in human melanoma. This locus encodes two distinct activated mutations in the N-ras gene has been documented. INK4a ARF ARF To obtain a mouse model most closely mimicking the human proteins by alternative exon usage, p16 and p14 (p19 in disease, a transgenic mouse line was generated by targeting mice), which function as tumor suppressor genes in the pRB and expression of dominant-active human N-ras (N-RasQ61K) to the p53 pathways, respectively (4, 5). Gene-targeted mice, where p16INK4a and p19ARF are deleted, develop a large variety of tumors melanocyte lineage by tyrosinase regulatory sequences INK4a Q61K but fail to develop melanoma (6). Mice deficient in p16 but (Tyr::N-Ras ). Transgenic mice show hyperpigmented skin ARF and develop cutaneous metastasizing melanoma. Consistent which retain one copy of p19 show carcinogen-induced with the tumor suppressor function of the INK4a locus that susceptibility to metastatic melanoma (7, 8). Transgenic mice that encodes p16INK4A and p19ARF, >90% of Tyr::N-RasQ61K INK4aÀ/À express a mutant form of H-ras specifically in melanocytes showed melanocytic hyperplasia with intense skin pigmentation (9), which transgenic mice develop melanoma at 6 months. -

Laser Treatment for Pigmentation
Laser treatment for pigmentation Sunspots and freckles In lesions such as solar lentigines (sun induced age spots), lentigo simplex and ephelides (freckles) the pigment (melanin) is in the top layers of the skin. Many lasers that selectively target melanin or simply remove the top layers will lead to improvement. One to two treatments will be necessary. There are many non-laser treatment approaches to treat such spots. However, if laser therapy is chosen, there are many different types of laser that can be used including Q-switched lasers [Q switched (QS) alexandrite, QS ruby (694nm)], long-pulsed, fractionated thulium (1927nm) and ablative lasers. IPL (filter 500-600nm) can also be very effective. However, it is common to see the pigmentation return over time. Treatments need to be avoided if the skin is tanned because this greatly increases the risk of complications. Seborrhoeic warts and dermatosis papulosa nigra Some conditions that present as dark spots are in fact wart-like and sit on the surface of the skin such as seborhoeic keratoses and dermatosis papulosa nigra and epidermal naevi. These conditions require physical removal. This is most commonly treated with a curette if thick or with fine wire diathermy particularly for the dermatosis papulosa nigra. Ablative lasers may also be used including CO2 in combination with a curette or an erbium YAG laser which reliably gives the best results. These lesions have a tendency to return over a number of years and maintenance treatments may be needed. Pigmented birth marks – Becker’s naevus Current lasers are unable to remove pigmented birthmarks without scarring. -

Dermatopathology
Dermatopathology Clay Cockerell • Martin C. Mihm Jr. • Brian J. Hall Cary Chisholm • Chad Jessup • Margaret Merola With contributions from: Jerad M. Gardner • Talley Whang Dermatopathology Clinicopathological Correlations Clay Cockerell Cary Chisholm Department of Dermatology Department of Pathology and Dermatopathology University of Texas Southwestern Medical Center Central Texas Pathology Laboratory Dallas , TX Waco , TX USA USA Martin C. Mihm Jr. Chad Jessup Department of Dermatology Department of Dermatology Brigham and Women’s Hospital Tufts Medical Center Boston , MA Boston , MA USA USA Brian J. Hall Margaret Merola Department of Dermatology Department of Pathology University of Texas Southwestern Medical Center Brigham and Women’s Hospital Dallas , TX Boston , MA USA USA With contributions from: Jerad M. Gardner Talley Whang Department of Pathology and Dermatology Harvard Vanguard Medical Associates University of Arkansas for Medical Sciences Boston, MA Little Rock, AR USA USA ISBN 978-1-4471-5447-1 ISBN 978-1-4471-5448-8 (eBook) DOI 10.1007/978-1-4471-5448-8 Springer London Heidelberg New York Dordrecht Library of Congress Control Number: 2013956345 © Springer-Verlag London 2014 This work is subject to copyright. All rights are reserved by the Publisher, whether the whole or part of the material is concerned, specifi cally the rights of translation, reprinting, reuse of illustrations, recitation, broadcasting, reproduction on microfi lms or in any other physical way, and transmission or information storage and retrieval, electronic adaptation, computer software, or by similar or dissimilar methodology now known or hereafter developed. Exempted from this legal reservation are brief excerpts in connection with reviews or scholarly analysis or material supplied specifi cally for the purpose of being entered and executed on a computer system, for exclusive use by the purchaser of the work. -
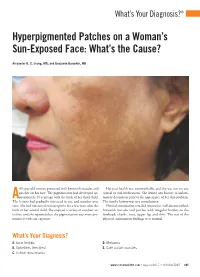
Hyperpigmented Patches on a Woman's Sun-Exposed Face
What’s Your Diagnosis?® Hyperpigmented Patches on a Woman’s Sun-Exposed Face: What’s the Cause? Alexander K. C. Leung, MD, and Benjamin Barankin, MD 45-year-old woman presented with brownish macules and Her past health was unremarkable, and she was not on any patches on her face. The pigmentation had developed ap- topical or oral medications. She denied any history of inflam- A proximately 10 years ago with the birth of her third child. matory dermatosis prior to the appearance of her skin problem. The lesions had gradually increased in size and number over The family history was not contributory. time. She had taken oral contraceptives for a few years after the Physical examination revealed symmetric, well-circumscribed, birth of her second child. She enjoyed a variety of outdoor ac- brownish macules and patches with irregular borders on the tivities, and she reported that the pigmentation was more pro- forehead, cheeks, nose, upper lip, and chin. The rest of the nounced with sun exposure. physical examination findings were normal. What’s Your Diagnosis? A. Solar lentigo D. Melasma B. Ephelides (freckles) E. Café au lait macules C. Actinic dyschromia www.consultant360.com • August 2017 • CONSULTANT 485 What’s Your Diagnosis?® Answer: Melasma A diagnosis of melasma was made. Wood lamp examination basement membrane disruption and dermal changes, indepen- showed accentuation of pigmentation suggestive of epidermal dent of UV radiation.1,17 pigmentation. The patient was treated with a series of trichlo- roacetic acid chemical peels, as well as a topical azelaic acid and HISTOPATHOLOGY a retinoid-hydroquinone-steroid (modified Kligman) formula- Three histologic patterns of pigmentation have been described: tion with significant, albeit incomplete, improvement. -

Quality of Life and Photodermatoses in People with Albinism in Benin City Nigeria
QUALITY OF LIFE AND PHOTODERMATOSES IN PEOPLE WITH ALBINISM IN BENIN CITY NIGERIA DISSERTATION SUBMITTED TO NATIONAL POSTGRADUATE MEDICAL COLLEGE OF NIGERIA IN PARTIAL FULFILMENT OF THE REQUIREMENTS FOR THE FELLOWSHIP IN INTERNAL MEDICINE (SUB-SPECIALTY: DERMATOLOGY) BY DR CYNTHIA ROLI MADUBUKO MB,BS (BENIN) DEPARTMENT OF MEDICINE UNIVERSITY OF BENIN TEACHING HOSPITAL, BENIN CITY, EDO STATE, NIGERIA NOVEMBER, 2016. i DECLARATION I hereby declare that this work is original and no part of it has been presented to any other college for a fellowship dissertation nor has it been submitted elsewhere for publication. Signature.................................... Date........................... Dr. Cynthia Roli Madubuko ii SUPERVISION We the undersigned have supervised the writing of this dissertation and the execution of this study. SIGNATURE: …………………………………… DATE: …………………………………………… YEAR OF FELLOWSHIP: ……………………… DR. B. OKWARA MBBS, FMCP Consultant Physician/ Dermatologist and Venereologist Department of Internal Medicine, University of Benin Teaching Hospital, Benin City, Nigeria. SIGNATURE: …………………………………… DATE: …………………………………………… YEAR OF FELLOWSHIP: ……………………… PROF. A.N. ONUNU MBBS, FWACP, FACP Consultant Physician/ Dermatologist and Venereologist Department of Internal Medicine, University of Benin Teaching Hospital, Benin City, Nigeria. SIGNATURE: …………………………………… DATE: …………………………………………… YEAR OF FELLOWSHIP: ……………………… PROF. E.P KUBEYINJE MBBS, FRCP (London), FWACP Consultant Physician Dermatologist and Venereologist Department of Internal Medicine, University of Benin Teaching Hospital, Benin City, Nigeria. iii CERTIFICATION I certify that this is a part 2 dissertation of the National postgraduate Medical College of Nigeria by Dr. Cynthia Roli Madubuko, of the Department of Internal Medicine, University of Benin Teaching Hospital, Benin City, under the supervision of Prof. A.N Onunu, Prof. E.P. Kubeyinje and Dr. B. Okwara Sign……………………………………. Date.............................................. DR OMUEMU, MBBS, FWACP. -

Dermamelan ® Treatment for Melasma and Hyperpigmentation
Dermamelan ® treatment for melasma and hyperpigmentation Jean Luc Levy Expert Laser, Dermatologist Lausanne, Switzerland [email protected] Incidence of Melasma Caucasian Melasma is estimated to occur in 50%-70% of pregnancies among US women, usually during the 2nd or 3rd trimester of pregnancy. Asian In Asian countries, the estimated prevalence of melasma within skin type III-V is estimated to be as high as 40% in females and 20% in males. Hispanic Among Mexican women, the incidence is estimated to be about 80%, with more than one-third of these patients having the disease for life. Retrospective Study on the Clinical Presentation and Treatment Outcome of Melasma in a Tertiary Dermatolofical Referral Centre in Singapore C L Goh, C N Dlova, Singapore Med J 1999; Vol 40(07): Melasma in Orientals. Sivayathorn A. Clin Drug Invest 1995; 10 (Suppl 2):24-40. ‘Utilizing combination therapy to optimize melasma outcomes’, M, I, Rendon(2004), Journal of Drugs in Dermatology, Sept-Oct. Facial melasma : Sun history Majority of melasma are very sensitive to sun exposure and tanned during summer - Sunblock regurlaly applied are poorly effective - During winter quality of life is better Some of them are poorly sensitive to sun exposure and colour acquired is near from the surrounding skin - During winter color differences between melasma and surrounding skin is high and covermark required Upper forehead without due to the tissue Sun exposed and non sun exposed areas Facial melasma : locations Forehead : with commonly a lighten aspect if hair is used -

Q-Switched 1064/532 Nm Laser with Picosecond Pulse to Treat Benign Hyperpigmentations: a Single-Center Retrospective Study
applied sciences Article Q-Switched 1064/532 nm Laser with Picosecond Pulse to Treat Benign Hyperpigmentations: A Single-Center Retrospective Study Martina Silvestri 1,2, Luigi Bennardo 1,3 , Elena Zappia 1, Federica Tamburi 1, Norma Cameli 2, Giovanni Cannarozzo 4,* and Steven Paul Nisticò 1 1 Department of Health Sciences, Magna Graecia University, 88100 Catanzaro, Italy; [email protected] (M.S.); [email protected] (L.B.); [email protected] (E.Z.); [email protected] (F.T.); [email protected] (S.P.N.) 2 San Gallicano Dermatological Institute, IRCCS, 00100 Rome, Italy; [email protected] 3 Unit of Dermatology, Mariano Santo Hospital, 87100 Cosenza, Italy 4 Unit of Dermatology, Tor Vergata University, 00100 Rome, Italy * Correspondence: [email protected]; Tel.: +39-09613647195 Abstract: (1) Benign melanoses are a frequent issue in aesthetic dermatology. Solar lentigo, ephelides, café au lait spots, and other melanoses represent a cosmetic issue for a growing number of subjects. The Q-switched 1064/532-nanometer (nm) laser may be considered the gold standard for manage- ment of these aesthetic issues. A new generation of Q-switched lasers, capable of concentrating the energy pulse in the spectrum of hundreds of picoseconds, is emerging, promising better results than previous ones. In this paper, we report the use of a Q-switched laser with a picosecond pulse to Citation: Silvestri, M.; Bennardo, L.; manage hypermelanoses. (2) Methods: 36 patients seeking melanosis removal were retrospectively Zappia, E.; Tamburi, F.; Cameli, N.; enrolled at Magna Graecia University of Catanzaro. Treatment parameters, although variable, were Cannarozzo, G.; Nisticò, S.P. -

Pediatric Dermatology- Pigmented Lesions
Pediatric Dermatology- Pigmented Lesions OPTI-West/Western University of Health Sciences- Silver Falls Dermatology Presenters: Bryce Lynn Desmond, DO; Ben Perry, DO Contributions from: Lauren Boudreaux, DO; Stephanie Howerter, DO; Collin Blattner, DO; Karsten Johnson, DO Disclosures • We have no financial or conflicts of interest to report Melanocyte Basic Science • Neural crest origin • Migrate to epidermis, dermis, leptomeninges, retina, choroid, iris, mucous membrane epithelium, inner ear, cochlea, vestibular system • Embryology • First appearance at the end of the 1st trimester • Able to synthesize melanin at the beginning of the 2nd trimester • Ratio of melanocytes to basal cells is 1:10 in skin and 1:4 in hair • Equal numbers of melanocytes across different races • Type, number, size, dispersion, and degree of melanization of the melanosomes determines pigmentation Nevus of Ota • A.k.a. Nevus Fuscocoeruleus Ophthalmomaxillaris • Onset at birth (50-60%) or 2nd decade • Larger than mongolian spot, does not typically regress spontaneously • Often first 2 branches of trigeminal nerve • Other involved sites include ipsilateral sclera (~66%), tympanum (55%), nasal mucosa (30%). • ~50 cases of melanoma reported • Reported rates of malignant transformation, 0.5%-25% in Asian populations • Ocular melanoma of choroid, orbit, chiasma, meninges have been observed in patients with clinical ocular hyperpigmentation. • Acquired variation seen in primarily Chinese or Japanese adults is called Hori’s nevus • Tx: Q-switched ruby, alexandrite, and -

Partial Unilateral Lentiginoses
igmentar f P y D l o i a so n r r d u e de Lima Grynszpan et al., Pigmentary Disorders 2015, 2:4 r o J s Journal of Pigmentary Disorders DOI: 10.4172/2376-0427.1000175 ISSN: 2376-0427 Case Report Open Access Partial Unilateral Lentiginoses Rachel de Lima Grynszpan1, Joao Paulo Niemeyer Corbellini2, Beatriz Maria Gaiser1 and Marcia Ramos-e-Silva3* 1Dermatologist and Former Post-Graduation Student – Sector of Dermatology and Post-Graduation Course, HUCFF-UFRJ and School of Medicine, Federal University of Rio de Janeiro, Rio de Janeiro, Brazil 2Dermatologist and Supervisor of the General Dermatology and Photodermatology Out-Patient Clinics – Sector of Dermatology and Post-Graduation Course, HUCFF- UFRJ and School of Medicine, Federal University of Rio de Janeiro, Rio de Janeiro, Brazil 3Associate Professor and Chair – Sector of Dermatology and Post-Graduation Course, HUCFF-UFRJ, and School of Medicine, Federal University of Rio de Janeiro, Rio de Janeiro, Brazil Abstract The authors present a case of a rare pigmentation disorder, partial unilateral lentiginosis (PUL), which is characterized by multiple lentigines located in only one side of the body with histopathology of lentigine. Keywords: Lentigines; Partial unilateral lentiginosis; Pigmentation At dermatologic examination, small brownish macules were observed on the back of the right hand, right cervical region, right Introduction upper chest, right lower lip and right forehead up to the medial line, Partial unilateral lentiginosis (PUL) is a rare pigmentation disorder characterized by multiple lentigines located in only one side of the body, which histopathologically show typical finding of lentigine [1,2]. It is characterized by small, well delimited, hyperchromic macules, distributed in a limited area of the body, which can affect one or more dermatomes and stop at the median line. -

Clinical Pigmented Skin Lesions Nontest-June 11
Recognizing Melanocytic Lesions James E. Fitzpatrick, M.D. University of Colorado Health Sciences Center No conflicts of interest to report Pigmented Skin Lesions L Pigmented keratinocyte neoplasias – Solar lentigo – Seborrheic keratosis – Pigmented actinic keratosis (uncommon) L Melanocytic hyperactivity – Ephelides (freckles) – Café-au-lait macules L Melanocytic neoplasia – Simple lentigo (lentigo simplex) – Benign nevocellular nevi – Dermal melanocytoses – Atypical (dysplastic) nevus – Malignant melanocytic lesions Solar Lentigo (Lentigo Senilis, Lentigo Solaris, Liver Spot, Age Spot) L Proliferation of keratinocytes with ↑ melanin – Variable hyperplasia in number of melanocytes L Pathogenesis- ultraviolet light damage Note associated solar purpura Solar Lentigo L Older patients L Light skin type L Photodistributed L Benign course L Problem- distinguishing form lentigo maligna Seborrheic Keratosis “Barnacles of Aging” L Epithelial proliferation L Common- 89% of geriatric population L Pathogenesis unknown – Follicular tumor (best evidence) – FGFR3 mutations in a subset Seborrheic Keratosis Clinical Features L Distribution- trunk>head and neck>extremities L Primary lesion – Exophytic papule with velvety to verrucous surface- “stuck on appearance” – Color- white, gray, tan, brown, black L Complications- inflammation, pruritus, and simulation of cutaneous malignancy L Malignancy potential- none to low (BCC?) Seborrheic Keratosis Seborrheic Keratosis- skin tag-like variant Pigmented Seborrheic Keratosis Inflamed Seborrheic Keratosis Café-au-Lait -

Lentigo Simplex= النمشة البسيطة
اﻠﺒﺴﻴﻄﺔ اﻠﻨﻤﺸﺔ =Lentigo simplex LENTIGO SIMPLEX Epidemiology The frequency of lentigo simplex in children and adults has not been determined. There does not appear to be a racial or gender predilection. Lentigo simplex is the most common histopathologic pattern of darkly pigmented lesions excised from acral sites of darkly pigmented races. Pigmented nail bands, noted in up to 20 percent of Japanese, may have histopathologic features of lentigo simplex. For agminated lentigines, the population prevalence is unknown, but it is believed to be rare. 1 / 8 اﻠﺒﺴﻴﻄﺔ اﻠﻨﻤﺸﺔ =Lentigo simplex Etiology and Pathogenesis The increased density of melanocytes in lentigines is presumably due to an underlying developmental or intrinsic defect in melanocyte homeostasis. In some lentigines, the presence of melanin macroglobules in melanocytes and keratinocytes suggests that defects affecting melanization pathways are also involved. The presence of lentigo simplex in association with somatic abnormalities in such diverse conditions as Peutz-Jeghers syndrome, LEPOARD syndrome (lentigines; electrocardiogram conduction defects; ocular hypertelorism; pulmonary stenosis; abnormalities of genitalia; retardation of growth, and sensorineural deafness), and LAMB/myxoma syndrome suggests that lentigo development may be influenced by a number of different genetic factors. For agminated lentigines, it is possible that chromosomal mosaicism plays a role in the development of the localized collection of lentigines. Clinical Findings HISTORY Unlike solar lentigines, lentigo simplex is thought to be largely UVR independent. Lentigo simplex may appear as early as the first decade and may occur anywhere on skin or mucous membranes. Generalized lentigines may occur as an isolated phenomenon without known familial aggregation and first appear at birth, during infancy, or during adulthood. -

Photoageing Skin of the Elderly
Malaysian Family Physician 2010; Volume 5, Number 1 ISSN: 1985-207X (print), 1985-2274 (electronic) ©Academy of Family Physicians of Malaysia Online version: http://www.e-mfp.org/ CME Article PHOTOAGEING SKIN OF THE ELDERLY SB Khoo FRACGP, Penang Medical College Address for correspondence: Dr Khoo Siew Beng, Senior Lecturer & Family Physician, Penang Medical College, No 4, Sepoy Lines Road, 10450 Penang, Malaysia. Tel: 604-2263 459, Email: [email protected] Khoo SB. Photoageing skin of the elderly. Malaysian Family Physician. 2010;5(1):9-12 CASE HISTORY Figure 2: Dorsal view of both forearms Mr A is an 80 year old man who presents with several warty skin lesions on his forearms for past 6 months. There was no complaint of pain or itch except occasional irritation when he accidentally rubbed against them. He noticed that these skin lesions had gradually increased in size and number. Mr A is generally well and healthy with a functional age of 65 years. He does not have any medical problem except for mild hypertension controlled well with atenolol 100mg daily. He used to work as a Chinese opera singer in his younger days and is now retired. Currently he spends his time providing service in church and help to make handicrafts to raise funds for the church and charitable old folk’s homes. Figure 3: Closer dorsal view of right forearm Physical examination (Figures 1-3) reveals extensive solar damaged skin on sun exposed areas of both forearms. There are scattered areas of hyperpigmentation (solar lentigo), isolated patches of hypopigmentation (solar hypomelanosis), skin atrophy, presence of wrinkles, telangiectasia and superficial areas of ecchymosis.