Sensitivity of Small-Cell Lung Cancer to BET Inhibition Is Mediated by Regulation of ASCL1 Gene Expression
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Screening of a Clinically and Biochemically Diagnosed SOD Patient Using Exome Sequencing: a Case Report with a Mutations/Variations Analysis Approach
The Egyptian Journal of Medical Human Genetics (2016) 17, 131–136 HOSTED BY Ain Shams University The Egyptian Journal of Medical Human Genetics www.ejmhg.eg.net www.sciencedirect.com CASE REPORT Screening of a clinically and biochemically diagnosed SOD patient using exome sequencing: A case report with a mutations/variations analysis approach Mohamad-Reza Aghanoori a,b,1, Ghazaleh Mohammadzadeh Shahriary c,2, Mahdi Safarpour d,3, Ahmad Ebrahimi d,* a Department of Medical Genetics, Shiraz University of Medical Sciences, Shiraz, Iran b Research and Development Division, RoyaBioGene Co., Tehran, Iran c Department of Genetics, Shahid Chamran University of Ahvaz, Ahvaz, Iran d Cellular and Molecular Research Center, Research Institute for Endocrine Sciences, Shahid Beheshti University of Medical Sciences, Tehran, Iran Received 12 May 2015; accepted 15 June 2015 Available online 22 July 2015 KEYWORDS Abstract Background: Sulfite oxidase deficiency (SOD) is a rare neurometabolic inherited disor- Sulfite oxidase deficiency; der causing severe delay in developmental stages and premature death. The disease follows an auto- Case report; somal recessive pattern of inheritance and causes deficiency in the activity of sulfite oxidase, an Exome sequencing enzyme that normally catalyzes conversion of sulfite to sulfate. Aim of the study: SOD is an underdiagnosed disorder and its diagnosis can be difficult in young infants as early clinical features and neuroimaging changes may imitate some common diseases. Since the prognosis of the disease is poor, using exome sequencing as a powerful and efficient strat- egy for identifying the genes underlying rare mendelian disorders can provide important knowledge about early diagnosis, disease mechanisms, biological pathways, and potential therapeutic targets. -

Role of Amylase in Ovarian Cancer Mai Mohamed University of South Florida, [email protected]
University of South Florida Scholar Commons Graduate Theses and Dissertations Graduate School July 2017 Role of Amylase in Ovarian Cancer Mai Mohamed University of South Florida, [email protected] Follow this and additional works at: http://scholarcommons.usf.edu/etd Part of the Pathology Commons Scholar Commons Citation Mohamed, Mai, "Role of Amylase in Ovarian Cancer" (2017). Graduate Theses and Dissertations. http://scholarcommons.usf.edu/etd/6907 This Dissertation is brought to you for free and open access by the Graduate School at Scholar Commons. It has been accepted for inclusion in Graduate Theses and Dissertations by an authorized administrator of Scholar Commons. For more information, please contact [email protected]. Role of Amylase in Ovarian Cancer by Mai Mohamed A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy Department of Pathology and Cell Biology Morsani College of Medicine University of South Florida Major Professor: Patricia Kruk, Ph.D. Paula C. Bickford, Ph.D. Meera Nanjundan, Ph.D. Marzenna Wiranowska, Ph.D. Lauri Wright, Ph.D. Date of Approval: June 29, 2017 Keywords: ovarian cancer, amylase, computational analyses, glycocalyx, cellular invasion Copyright © 2017, Mai Mohamed Dedication This dissertation is dedicated to my parents, Ahmed and Fatma, who have always stressed the importance of education, and, throughout my education, have been my strongest source of encouragement and support. They always believed in me and I am eternally grateful to them. I would also like to thank my brothers, Mohamed and Hussien, and my sister, Mariam. I would also like to thank my husband, Ahmed. -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

Biological Models of Colorectal Cancer Metastasis and Tumor Suppression
BIOLOGICAL MODELS OF COLORECTAL CANCER METASTASIS AND TUMOR SUPPRESSION PROVIDE MECHANISTIC INSIGHTS TO GUIDE PERSONALIZED CARE OF THE COLORECTAL CANCER PATIENT By Jesse Joshua Smith Dissertation Submitted to the Faculty of the Graduate School of Vanderbilt University In partial fulfillment of the requirements For the degree of DOCTOR OF PHILOSOPHY In Cell and Developmental Biology May, 2010 Nashville, Tennessee Approved: Professor R. Daniel Beauchamp Professor Robert J. Coffey Professor Mark deCaestecker Professor Ethan Lee Professor Steven K. Hanks Copyright 2010 by Jesse Joshua Smith All Rights Reserved To my grandparents, Gladys and A.L. Lyth and Juanda Ruth and J.E. Smith, fully supportive and never in doubt. To my amazing and enduring parents, Rebecca Lyth and Jesse E. Smith, Jr., always there for me. .my sure foundation. To Jeannine, Bill and Reagan for encouragement, patience, love, trust and a solid backing. To Granny George and Shawn for loving support and care. And To my beautiful wife, Kelly, My heart, soul and great love, Infinitely supportive, patient and graceful. ii ACKNOWLEDGEMENTS This work would not have been possible without the financial support of the Vanderbilt Medical Scientist Training Program through the Clinical and Translational Science Award (Clinical Investigator Track), the Society of University Surgeons-Ethicon Scholarship Fund and the Surgical Oncology T32 grant and the Vanderbilt Medical Center Section of Surgical Sciences and the Department of Surgical Oncology. I am especially indebted to Drs. R. Daniel Beauchamp, Chairman of the Section of Surgical Sciences, Dr. James R. Goldenring, Vice Chairman of Research of the Department of Surgery, Dr. Naji N. -

Supplemental Figure S1 Differentially Methylated Regions (Dmrs
Supplemental Figure S1 '$$#0#,2'**7+#2&7*2#"0#%'-,11 #25##,"'1#1#122#1 '!2-0'*"#.'!2'-,-$122,1'2'-,$0-+2- !"Q !"2-$%," $ 31',% 25-$-*" !&,%# ," ' 0RTRW 1 !32V-$$ !0'2#0'T - #.0#1#,22'-, -$ "'$$#0#,2'**7+#2&7*2#"%#,#11',.0#,2#1,"2&#'0 #&4'-022,1'2'-, #25##,"'$$#0#,2"'1#1#122#1T-*!)00-51',"'!2#&7.#0+#2&7*2#"%#,#1Q%0700-51 &7.-+#2&7*2#"%#,#1Q31',%25-$-*"!&,%#,"'0RTRW1!32V-$$!0'2#0'T-%#,#1 +#22&# -4#!0'2#0'22,1'2'-,$0-+$%2-$Q5#2�#$-0#*1-',!*3"#" %#,#15'2&V4*3#0RTRWT$$#!2#"%#,10#&'%&*'%&2#" 712#0'1)1#T Supplemental Figure S2 Validation of results from the HELP assay using Epityper MassarrayT #13*21 $0-+ 2&# 1$ 117 5#0# !-00#*2#" 5'2& /3,2'22'4# +#2&7*2'-, ,*78#" 7 '13*$'2#11007$-04V-,"6U-%#,#.0-+-2#00#%'-,1T11007 51.#0$-0+#"31',%**4'* *#1+.*#1T S Supplemental Fig. S1 A unique hypermethylated genes (methylation sites) 454 (481) 5693 (6747) 120 (122) NLMGUS NEWMM REL 2963 (3207) 1338 (1560) 5 (5) unique hypomethylated genes (methylation sites) B NEWMM 0 (0) MGUS 454 (481) 0 (0) NEWMM REL NL 3* (2) 2472 (3066) NEWMM 2963 REL (3207) 2* (2) MGUS 0 (0) REL 2 (2) NEWMM 0 (0) REL Supplemental Fig. S2 A B ARID4B DNMT3A Methylation by MassArray Methylation by MassArray 0 0.2 0.4 0.6 0.8 1 1.2 0.5 0.6 0.7 0.8 0.9 1 2 0 NL PC MGUS 1.5 -0.5 NEW MM 1 REL MM -1 0.5 -1.5 0 -2 -0.5 -1 -2.5 -1.5 -3 Methylation by HELP Assay Methylation by HELP Methylation by HELP Assay Methylation by HELP -2 -3.5 -2.5 -4 Supplemental tables "3..*#+#,2*6 *#"SS 9*','!*!&0!2#0'12'!1-$.2'#,21+.*#1 DZ_STAGE Age Gender Ethnicity MM isotype PCLI Cytogenetics -

Mai Muudatuntuu Ti on Man Mini
MAIMUUDATUNTUU US009809854B2 TI ON MAN MINI (12 ) United States Patent ( 10 ) Patent No. : US 9 ,809 ,854 B2 Crow et al. (45 ) Date of Patent : Nov . 7 , 2017 Whitehead et al. (2005 ) Variation in tissue - specific gene expression ( 54 ) BIOMARKERS FOR DISEASE ACTIVITY among natural populations. Genome Biology, 6 :R13 . * AND CLINICAL MANIFESTATIONS Villanueva et al. ( 2011 ) Netting Neutrophils Induce Endothelial SYSTEMIC LUPUS ERYTHEMATOSUS Damage , Infiltrate Tissues, and Expose Immunostimulatory Mol ecules in Systemic Lupus Erythematosus . The Journal of Immunol @(71 ) Applicant: NEW YORK SOCIETY FOR THE ogy , 187 : 538 - 552 . * RUPTURED AND CRIPPLED Bijl et al. (2001 ) Fas expression on peripheral blood lymphocytes in MAINTAINING THE HOSPITAL , systemic lupus erythematosus ( SLE ) : relation to lymphocyte acti vation and disease activity . Lupus, 10 :866 - 872 . * New York , NY (US ) Crow et al . (2003 ) Microarray analysis of gene expression in lupus. Arthritis Research and Therapy , 5 :279 - 287 . * @(72 ) Inventors : Mary K . Crow , New York , NY (US ) ; Baechler et al . ( 2003 ) Interferon - inducible gene expression signa Mikhail Olferiev , Mount Kisco , NY ture in peripheral blood cells of patients with severe lupus . PNAS , (US ) 100 ( 5 ) : 2610 - 2615. * GeneCards database entry for IFIT3 ( obtained from < http : / /www . ( 73 ) Assignee : NEW YORK SOCIETY FOR THE genecards. org /cgi - bin / carddisp .pl ? gene = IFIT3 > on May 26 , 2016 , RUPTURED AND CRIPPLED 15 pages ) . * Navarra et al. (2011 ) Efficacy and safety of belimumab in patients MAINTAINING THE HOSPITAL with active systemic lupus erythematosus : a randomised , placebo FOR SPECIAL SURGERY , New controlled , phase 3 trial . The Lancet , 377 :721 - 731. * York , NY (US ) Abramson et al . ( 1983 ) Arthritis Rheum . -

Application of Microrna Database Mining in Biomarker Discovery and Identification of Therapeutic Targets for Complex Disease
Article Application of microRNA Database Mining in Biomarker Discovery and Identification of Therapeutic Targets for Complex Disease Jennifer L. Major, Rushita A. Bagchi * and Julie Pires da Silva * Department of Medicine, Division of Cardiology, University of Colorado Anschutz Medical Campus, Aurora, CO 80045, USA; [email protected] * Correspondence: [email protected] (R.A.B.); [email protected] (J.P.d.S.) Supplementary Tables Methods Protoc. 2021, 4, 5. https://doi.org/10.3390/mps4010005 www.mdpi.com/journal/mps Methods Protoc. 2021, 4, 5. https://doi.org/10.3390/mps4010005 2 of 25 Table 1. List of all hsa-miRs identified by Human microRNA Disease Database (HMDD; v3.2) analysis. hsa-miRs were identified using the term “genetics” and “circulating” as input in HMDD. Targets CAD hsa-miR-1 Targets IR injury hsa-miR-423 Targets Obesity hsa-miR-499 hsa-miR-146a Circulating Obesity Genetics CAD hsa-miR-423 hsa-miR-146a Circulating CAD hsa-miR-149 hsa-miR-499 Circulating IR Injury hsa-miR-146a Circulating Obesity hsa-miR-122 Genetics Stroke Circulating CAD hsa-miR-122 Circulating Stroke hsa-miR-122 Genetics Obesity Circulating Stroke hsa-miR-26b hsa-miR-17 hsa-miR-223 Targets CAD hsa-miR-340 hsa-miR-34a hsa-miR-92a hsa-miR-126 Circulating Obesity Targets IR injury hsa-miR-21 hsa-miR-423 hsa-miR-126 hsa-miR-143 Targets Obesity hsa-miR-21 hsa-miR-223 hsa-miR-34a hsa-miR-17 Targets CAD hsa-miR-223 hsa-miR-92a hsa-miR-126 Targets IR injury hsa-miR-155 hsa-miR-21 Circulating CAD hsa-miR-126 hsa-miR-145 hsa-miR-21 Targets Obesity hsa-mir-223 hsa-mir-499 hsa-mir-574 Targets IR injury hsa-mir-21 Circulating IR injury Targets Obesity hsa-mir-21 Targets CAD hsa-mir-22 hsa-mir-133a Targets IR injury hsa-mir-155 hsa-mir-21 Circulating Stroke hsa-mir-145 hsa-mir-146b Targets Obesity hsa-mir-21 hsa-mir-29b Methods Protoc. -
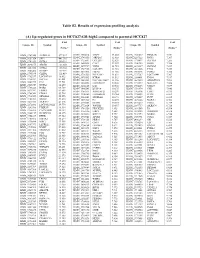
(A) Up-Regulated Genes in HCC827-GR-High2 Compared to Parental HCC827
Table S2. Results of expression profiling analysis (A) Up-regulated genes in HCC827-GR-high2 compared to parental HCC827 Fold Fold Fold Unique ID Symbol Unique ID Symbol Unique ID Symbol change* change* change* ILMN_1709348 ALDH1A1 577.587 ILMN_2310814 MAPT 13.003 ILMN_1741017 PIP4K2B 7.331 ILMN_1651354 SPP1 441.316 ILMN_1748650 MRPL45 12.988 ILMN_3237623 RNY1 7.297 ILMN_1701831 GSTA1 260.591 ILMN_1755897 UGT2B7 12.629 ILMN_1734897 SLC4A4 7.285 ILMN_1658835 CAV2 12.357 ILMN_1746359 RERG 7.280 ILMN_2094875 ABCB1 183.050 ILMN_1678939 VNN2 11.935 ILMN_1671337 SLC2A5 7.257 ILMN_3251540 GSTA2 145.982 ILMN_1729905 GAL3ST1 11.910 ILMN_1691606 LYG2 7.254 ILMN_2062468 IGFBP7 127.721 ILMN_1672536 FBLN1 11.716 ILMN_1785646 PMP22 7.246 ILMN_1795190 CLDN2 111.439 ILMN_1796339 PLEKHA2 11.631 ILMN_1737387 LOC728441 7.207 ILMN_1782937 LOC647169 98.612 ILMN_1676563 HTRA1 11.592 ILMN_1684401 FMO1 7.117 ILMN_1754247 SLC3A1 81.001 ILMN_3263423 LOC100129027 11.346 ILMN_1687035 ADAMTSL4 7.098 ILMN_1662795 CA2 79.581 ILMN_1694898 LOC653857 10.906 ILMN_2153572 MAGEA3 7.086 ILMN_2168747 GSTA2 66.250 ILMN_2404625 LAT 10.560 ILMN_1784283 USH1C 7.079 ILMN_1764228 DAB2 64.709 ILMN_1666546 DUSP14 10.375 ILMN_1731374 CPE 7.046 ILMN_1675797 EPDR1 63.605 ILMN_1764571 ARHGAP23 10.299 ILMN_1765446 EMP3 6.933 ILMN_1708341 PDZK1 59.714 ILMN_3200140 LOC645638 10.284 ILMN_1754002 IL1F8 6.863 ILMN_1713529 SEMA6A 52.575 ILMN_3244343 SNORA21 10.171 ILMN_1878007 FUT9 6.835 ILMN_1708391 NR1H4 43.218 ILMN_1671489 PC 10.075 ILMN_1699208 NAP1L1 6.763 ILMN_2412336 AKR1C2 42.826 ILMN_2404688 -

Atlas of the Open Reading Frames in Human Diseases: Dark Matter of the Human Genome
MOJ Proteomics & Bioinformatics Research Article Open Access Atlas of the open reading frames in human diseases: dark matter of the human genome Abstract Volume 2 Issue 1 - 2015 Background: The human genome encodes RNA and protein coding sequences, the Ana Paula Delgado, Maria Julia Chapado, non-coding RNAs, pseudogenes and uncharacterized Open Reading Frames (ORFs). The dark matter of the human genome encompassing the uncharacterized proteins and Pamela Brandao, Sheilin Hamid, Ramaswamy the non-coding RNAs are least well studied. However, they offer novel druggable Narayanan targets and biomarkers discovery for diverse diseases. Department of Biological Sciences, Charles E. Schmidt College of Science, Florida Atlantic University, USA Methods: In this study, we have systematically dissected the dark matter of the human genome. Using diverse bioinformatics tools, an atlas of the dark matter of the genome Correspondence: Ramaswamy Narayanan, Department of was created. The ORFs were characterized for gene ontology, mRNA and protein Biological Sciences, Charles E. Schmidt College of Science, expression, protein motif and domains and genome-wide association. Florida Atlantic University, FL 33431, USA. Tel +15612972247, Fax +15612973859, Email [email protected] Results: An atlas of disease-related ORFs (n=800) was generated. A complex landscape of involvement in multiple diseases was seen for these ORFs. Motif and Received: January 08, 2015 | Published: January 30, 2015 domains analysis identified druggable targets and putative secreted biomarkers including enzymes, receptors and transporters as well as proteins with signal peptide sequence. About ten percent of the ORFs showed a correlation of gene expression at the mRNA and protein levels. Genome-Phenome association tools identified ORF’s association with autoimmune, cardiovascular, cancer, diabetes, infection, metabolic, neurodegenerative and psychiatric disorders. -

Biomedical Robots. Application to Translational Medicine
Biomedical robots. Application to translational medicine. Enrique J. deAndrés-Galiana Supervisors: Prof. Juan Luis Fernández-Martínez & Prof. Oscar Luaces This dissertation is submitted under the PhD program of Mathematics and Statistics May 2016 RESUMEN DEL CONTENIDO DE TESIS DOCTORAL 1.- Título de la Tesis Español/Otro Idioma: Inglés: Diseño de robots biomédicos. Aplicaciones en Biomedical robots. Application to translational medicina traslacional. medicine. 2.- Autor Nombre: Enrique Juan de Andrés Galiana DNI/Pasaporte/NIE: Programa de Doctorado: Matemáticas y Estadística. Órgano responsable: Departamento de Matemáticas. RESUMEN (en español) Esta tesis trata sobre el análisis y diseño de robots biomédicos y su aplicación a la medicina traslacional. Se define un robot biomédico como el conjunto de técnicas provenientes de la matemática aplicada, estadística y ciencias de la computación capaces de analizar datos biomédicos de alta dimensionalidad, aprender dinámicamente de dichos datos, extraer nuevo BIS - conocimiento e hipótesis de trabajo, y finalmente realizar predicciones con su incertidumbre asociada, cara a la toma de decisiones biomédicas. Se diseñan y analizan diferentes algorit- 010 - mos de aprendizaje, de reducción de la dimensión y selección de atributos, así como técnicas de optimización global, técnicas de agrupamiento no supervisado, clasificación y análisis de VOA incertidumbre. Dichas metodologías se aplican a datos a pie de hospital y de expresión génica - en predicción de fenotipos para optimización del diagnóstico, pronóstico, tratamiento y análisis de toxicidades. MAT - Se muestra que es posible establecer de modo sencillo el poder discriminatorio de las variables FOR pronóstico, y que dichos problemas de clasificación se aproximan a un comportamiento linealmente separable cuando se reduce la dimensión al conjunto de variables principales que definen el alfabeto del problema biomédico y están por tanto relacionadas con su génesis. -

The Neuroprotective Role of the GM1 Oligosaccharide, Ii3neu5ac-Gg4, In
Molecular Neurobiology (2019) 56:6673–6702 https://doi.org/10.1007/s12035-019-1556-8 The Neuroprotective Role of the GM1 Oligosaccharide, 3 II Neu5Ac-Gg4, in Neuroblastoma Cells Elena Chiricozzi1 & Margherita Maggioni1 & Erika di Biase1 & Giulia Lunghi1 & Maria Fazzari1 & Nicoletta Loberto 1 & Maffioli Elisa2 & Francesca Grassi Scalvini2 & Gabriella Tedeschi 2,3 & Sandro Sonnino1 Received: 10 January 2019 /Accepted: 13 March 2019 /Published online: 26 March 2019 # Springer Science+Business Media, LLC, part of Springer Nature 2019 Abstract 3 Recently, we demonstrated that the GM1 oligosaccharide, II Neu5Ac-Gg4 (OligoGM1), administered to cultured murine Neuro2a neuroblastoma cells interacts with the NGF receptor TrkA, leading to the activation of the ERK1/2 downstream pathway and to cell differentiation. To understand how the activation of the TrkA pathway is able to trigger key biochemical signaling, we performed a proteomic analysis on Neuro2a cells treated with 50 μM OligoGM1 for 24 h. Over 3000 proteins were identified. Among these, 324 proteins were exclusively expressed in OligoGM1-treated cells. Interestingly, several proteins expressed only in OligoGM1-treated cells are involved in biochemical mechanisms with a neuroprotective potential, reflecting the GM1 neuroprotective effect. In addition, we found that the exogenous administration of OligoGM1 reduced the cellular oxidative stress in Neuro2a cells and conferred protection against MPTP neurotoxicity. These results confirm and reinforce the idea that the molecular mechanisms underlying the GM1 neurotrophic and neuroprotective effects depend on its oligosaccharide chain, suggesting the activation of a positive signaling starting at plasma membrane level. Keywords GM1 ganglioside . GM1 oligosaccharide chain . TrkA neurotrophin receptor . Plasma membrane signaling . Neuroprotection . -

Screening of a Clinically and Biochemically Diagnosed SOD Patient Using Exome Sequencing: a Case Report with a Mutations/Variations Analysis Approach
The Egyptian Journal of Medical Human Genetics (2016) 17, 131–136 HOSTED BY Ain Shams University The Egyptian Journal of Medical Human Genetics www.ejmhg.eg.net www.sciencedirect.com CASE REPORT Screening of a clinically and biochemically diagnosed SOD patient using exome sequencing: A case report with a mutations/variations analysis approach Mohamad-Reza Aghanoori a,b,1, Ghazaleh Mohammadzadeh Shahriary c,2, Mahdi Safarpour d,3, Ahmad Ebrahimi d,* a Department of Medical Genetics, Shiraz University of Medical Sciences, Shiraz, Iran b Research and Development Division, RoyaBioGene Co., Tehran, Iran c Department of Genetics, Shahid Chamran University of Ahvaz, Ahvaz, Iran d Cellular and Molecular Research Center, Research Institute for Endocrine Sciences, Shahid Beheshti University of Medical Sciences, Tehran, Iran Received 12 May 2015; accepted 15 June 2015 Available online 22 July 2015 KEYWORDS Abstract Background: Sulfite oxidase deficiency (SOD) is a rare neurometabolic inherited disor- Sulfite oxidase deficiency; der causing severe delay in developmental stages and premature death. The disease follows an auto- Case report; somal recessive pattern of inheritance and causes deficiency in the activity of sulfite oxidase, an Exome sequencing enzyme that normally catalyzes conversion of sulfite to sulfate. Aim of the study: SOD is an underdiagnosed disorder and its diagnosis can be difficult in young infants as early clinical features and neuroimaging changes may imitate some common diseases. Since the prognosis of the disease is poor, using exome sequencing as a powerful and efficient strat- egy for identifying the genes underlying rare mendelian disorders can provide important knowledge about early diagnosis, disease mechanisms, biological pathways, and potential therapeutic targets.