NAP1L4 Polyclonal Antibody Purified Rabbit Polyclonal Antibody (Pab) Catalog # AP57353
Total Page:16
File Type:pdf, Size:1020Kb
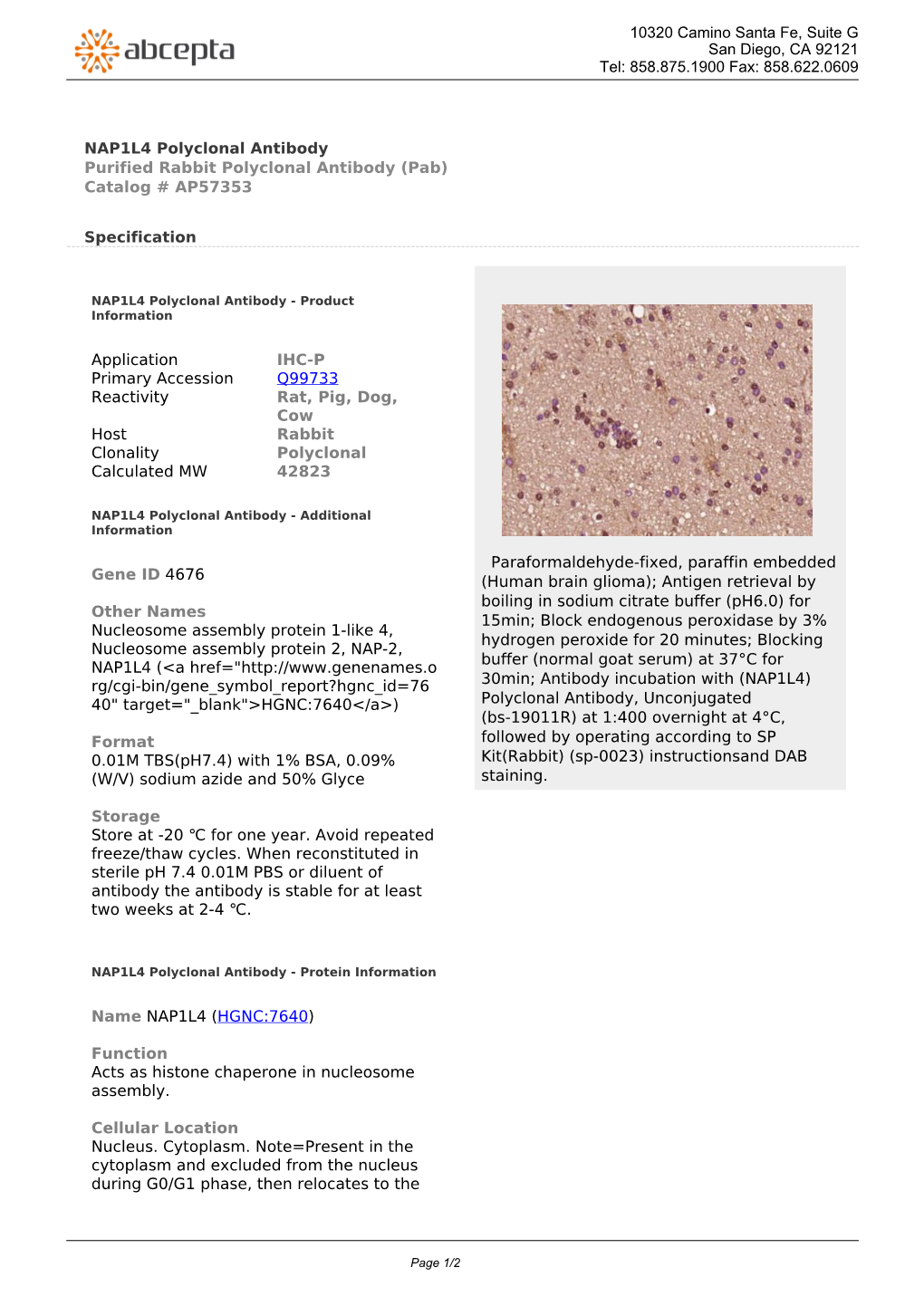
Load more
Recommended publications
-

Aberrant Methylation Underlies Insulin Gene Expression in Human Insulinoma
ARTICLE https://doi.org/10.1038/s41467-020-18839-1 OPEN Aberrant methylation underlies insulin gene expression in human insulinoma Esra Karakose1,6, Huan Wang 2,6, William Inabnet1, Rajesh V. Thakker 3, Steven Libutti4, Gustavo Fernandez-Ranvier 1, Hyunsuk Suh1, Mark Stevenson 3, Yayoi Kinoshita1, Michael Donovan1, Yevgeniy Antipin1,2, Yan Li5, Xiaoxiao Liu 5, Fulai Jin 5, Peng Wang 1, Andrew Uzilov 1,2, ✉ Carmen Argmann 1, Eric E. Schadt 1,2, Andrew F. Stewart 1,7 , Donald K. Scott 1,7 & Luca Lambertini 1,6 1234567890():,; Human insulinomas are rare, benign, slowly proliferating, insulin-producing beta cell tumors that provide a molecular “recipe” or “roadmap” for pathways that control human beta cell regeneration. An earlier study revealed abnormal methylation in the imprinted p15.5-p15.4 region of chromosome 11, known to be abnormally methylated in another disorder of expanded beta cell mass and function: the focal variant of congenital hyperinsulinism. Here, we compare deep DNA methylome sequencing on 19 human insulinomas, and five sets of normal beta cells. We find a remarkably consistent, abnormal methylation pattern in insu- linomas. The findings suggest that abnormal insulin (INS) promoter methylation and altered transcription factor expression create alternative drivers of INS expression, replacing cano- nical PDX1-driven beta cell specification with a pathological, looping, distal enhancer-based form of transcriptional regulation. Finally, NFaT transcription factors, rather than the cano- nical PDX1 enhancer complex, are predicted to drive INS transactivation. 1 From the Diabetes Obesity and Metabolism Institute, The Department of Surgery, The Department of Pathology, The Department of Genetics and Genomics Sciences and The Institute for Genomics and Multiscale Biology, The Icahn School of Medicine at Mount Sinai, New York, NY 10029, USA. -

Research Article Characterization, Tissue Expression, and Imprinting Analysis of the Porcine CDKN1C and NAP1L4 Genes
Hindawi Publishing Corporation Journal of Biomedicine and Biotechnology Volume 2012, Article ID 946527, 7 pages doi:10.1155/2012/946527 Research Article Characterization, Tissue Expression, and Imprinting Analysis of the Porcine CDKN1C and NAP1L4 Genes Shun Li,1 Juan Li,1 Jiawei Tian,1 Ranran Dong,1 Jin Wei,1 Xiaoyan Qiu,2 and Caode Jiang2 1 School of Life Science, Southwest University, Chongqing 400715, China 2 College of Animal Science and Technology, Southwest University, Chongqing 400715, China Correspondence should be addressed to Caode Jiang, [email protected] Received 4 August 2011; Revised 25 October 2011; Accepted 15 November 2011 Academic Editor: Andre Van Wijnen Copyright © 2012 Shun Li et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. CDKN1C and NAP1L4 in human CDKN1C/KCNQ1OT1 imprinted domain are two key candidate genes responsible for BWS (Beckwith-Wiedemann syndrome) and cancer. In order to increase understanding of these genes in pigs, their cDNAs are characterized in this paper. By the IMpRH panel, porcine CDKN1C and NAP1L4 genes were assigned to porcine chromosome 2, closely linked with IMpRH06175 and with LOD of 15.78 and 17.94, respectively. By real-time quantitative RT-PCR and polymorphism-based method, tissue and allelic expression of both genes were determined using F1 pigs of Rongchang and Landrace reciprocal crosses. The transcription levels of porcine CDKN1C and NAP1L4 were significantly higher in placenta than in other neonatal tissues (P<0.01) although both genes showed the highest expression levels in the lung and kidney of one- month pigs (P<0.01). -

Snps) Distant from Xenobiotic Response Elements Can Modulate Aryl Hydrocarbon Receptor Function: SNP-Dependent CYP1A1 Induction S
Supplemental material to this article can be found at: http://dmd.aspetjournals.org/content/suppl/2018/07/06/dmd.118.082164.DC1 1521-009X/46/9/1372–1381$35.00 https://doi.org/10.1124/dmd.118.082164 DRUG METABOLISM AND DISPOSITION Drug Metab Dispos 46:1372–1381, September 2018 Copyright ª 2018 by The American Society for Pharmacology and Experimental Therapeutics Single Nucleotide Polymorphisms (SNPs) Distant from Xenobiotic Response Elements Can Modulate Aryl Hydrocarbon Receptor Function: SNP-Dependent CYP1A1 Induction s Duan Liu, Sisi Qin, Balmiki Ray,1 Krishna R. Kalari, Liewei Wang, and Richard M. Weinshilboum Division of Clinical Pharmacology, Department of Molecular Pharmacology and Experimental Therapeutics (D.L., S.Q., B.R., L.W., R.M.W.) and Division of Biomedical Statistics and Informatics, Department of Health Sciences Research (K.R.K.), Mayo Clinic, Rochester, Minnesota Received April 22, 2018; accepted June 28, 2018 ABSTRACT Downloaded from CYP1A1 expression can be upregulated by the ligand-activated aryl fashion. LCLs with the AA genotype displayed significantly higher hydrocarbon receptor (AHR). Based on prior observations with AHR-XRE binding and CYP1A1 mRNA expression after 3MC estrogen receptors and estrogen response elements, we tested treatment than did those with the GG genotype. Electrophoretic the hypothesis that single-nucleotide polymorphisms (SNPs) map- mobility shift assay (EMSA) showed that oligonucleotides with the ping hundreds of base pairs (bp) from xenobiotic response elements AA genotype displayed higher LCL nuclear extract binding after (XREs) might influence AHR binding and subsequent gene expres- 3MC treatment than did those with the GG genotype, and mass dmd.aspetjournals.org sion. -

Letters to the Editor J Med Genet: First Published As 10.1136/Jmg.37.3.231 on 1 March 2000
210 Letters Letters to the Editor J Med Genet: first published as 10.1136/jmg.37.3.231 on 1 March 2000. Downloaded from J Med Genet 2000;37:210–212 No evidence of germline PTEN three reasons: somatic mutations have been found in PTEN in prostate tumours; germline mutations in Cowden mutations in familial prostate cancer disease produce a phenotype (although with no evidence of an associated susceptibility to prostate cancer); and PTEN deficient mice exhibit prostate abnormalities. We have therefore screened the Cancer Research Campaign/British EDITOR—Prostate cancer is the second most common Prostate Group (CRC/BPG) UK Familial Prostate Cancer cause of male cancer mortality in the UK.1 Current indica- Study samples for evidence of PTEN mutations. tions are that like many common cancers, prostate cancer The CRC/BPG UK Familial Prostate Cancer Study has has an inherited component.2 Segregation analysis has led collected lymphocyte DNA from 188 subjects from 50 to the proposed model of at least one highly penetrant, prostate cancer families. These families were chosen dominant gene (with an estimated 88% penetrance for because each contained three or more cases of prostate prostate cancer by the age of 85 in the highly susceptible cancer at any age or related sib pairs where at least one man population). Such a gene or genes would account for an was less than 67 (original criterion was 65) years old at estimated 43% of cases diagnosed at less than 55 years.2 diagnosis. In fact, the majority of the clusters consist of One prostate cancer susceptibility locus (HPC1) has been aVected sib pairs, with DNA often only available from reported on 1q24-253 and confirmed by Cooney et al4 and cases. -

Mclean, Chelsea.Pdf
COMPUTATIONAL PREDICTION AND EXPERIMENTAL VALIDATION OF NOVEL MOUSE IMPRINTED GENES A Dissertation Presented to the Faculty of the Graduate School of Cornell University In Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy by Chelsea Marie McLean August 2009 © 2009 Chelsea Marie McLean COMPUTATIONAL PREDICTION AND EXPERIMENTAL VALIDATION OF NOVEL MOUSE IMPRINTED GENES Chelsea Marie McLean, Ph.D. Cornell University 2009 Epigenetic modifications, including DNA methylation and covalent modifications to histone tails, are major contributors to the regulation of gene expression. These changes are reversible, yet can be stably inherited, and may last for multiple generations without change to the underlying DNA sequence. Genomic imprinting results in expression from one of the two parental alleles and is one example of epigenetic control of gene expression. So far, 60 to 100 imprinted genes have been identified in the human and mouse genomes, respectively. Identification of additional imprinted genes has become increasingly important with the realization that imprinting defects are associated with complex disorders ranging from obesity to diabetes and behavioral disorders. Despite the importance imprinted genes play in human health, few studies have undertaken genome-wide searches for new imprinted genes. These have used empirical approaches, with some success. However, computational prediction of novel imprinted genes has recently come to the forefront. I have developed generalized linear models using data on a variety of sequence and epigenetic features within a training set of known imprinted genes. The resulting models were used to predict novel imprinted genes in the mouse genome. After imposing a stringency threshold, I compiled an initial candidate list of 155 genes. -

Is Associated with Hypomethylation At
797 ORIGINAL ARTICLE J Med Genet: first published as 10.1136/jmg.40.11.797 on 19 November 2003. Downloaded from Silencing of CDKN1C (p57KIP2) is associated with hypomethylation at KvDMR1 in Beckwith–Wiedemann syndrome N Diaz-Meyer, C D Day, K Khatod, E R Maher, W Cooper, W Reik, C Junien, G Graham, E Algar, V M Der Kaloustian, M J Higgins ............................................................................................................................... J Med Genet 2003;40:797–801 Context: Beckwith–Wiedemann syndrome (BWS) arises by several genetic and epigenetic mechanisms affecting the balance of imprinted gene expression in chromosome 11p15.5. The most frequent alteration associated with BWS is the absence of methylation at the maternal allele of KvDMR1, an intronic CpG island within the KCNQ1 gene. Targeted deletion of KvDMR1 suggests that this locus is an imprinting control region (ICR) that regulates multiple genes in 11p15.5. Cell culture based enhancer blocking assays indicate that KvDMR1 may function as a methylation modulated chromatin insulator and/or silencer. See end of article for Objective: To determine the potential consequence of loss of methylation (LOM) at KvDMR1 in the authors’ affiliations ....................... development of BWS. Methods: The steady state levels of CDKN1C gene expression in fibroblast cells from normal individuals, Correspondence to: and from persons with BWS who have LOM at KvDMR1, was determined by both real time quantitative M J Higgins, Department of Cancer Genetics, polymerase chain reaction (qPCR) and ribonuclease protection assay (RPA). Methylation of the CDKN1C Roswell Park Cancer promoter region was assessed by Southern hybridisation using a methylation sensitive restriction Institute, Buffalo, NY endonuclease. 14263, USA; michael.higgins@ Results: Both qPCR and RPA clearly demonstrated a marked decrease (86–93%) in the expression level of roswellpark.org the CDKN1C gene in cells derived from patients with BWS, who had LOM at KvDMR1. -

Associated with Spherical Equivalent
Molecular Vision 2016; 22:783-796 <http://www.molvis.org/molvis/v22/783> © 2016 Molecular Vision Received 20 August 2015 | Accepted 12 July 2016 | Published 14 July 2016 Variation in PTCHD2, CRISP3, NAP1L4, FSCB, and AP3B2 associated with spherical equivalent Fei Chen,1 Priya Duggal,1 Barbara E.K. Klein,2 Kristine E. Lee,2 Barbara Truitt,3 Ronald Klein,2 Sudha K. Iyengar,3 Alison P. Klein1,4,5 1Department of Epidemiology, Johns Hopkins Bloomberg School of Public Health, Baltimore, MD; 2Department of Ophthalmology and Visual Sciences, University of Wisconsin School of Medicine and Public Health, Madison, WI; 3Department of Epidemiology and Biostatistics, Case Western Reserve University, Cleveland, OH; 4Department of Oncology, Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins, Baltimore, MD; 5Department of Pathology, Johns Hopkins School of Medicine, Baltimore, MD Purpose: Ocular refraction is measured in spherical equivalent as the power of the external lens required to focus images on the retina. Myopia (nearsightedness) and hyperopia (farsightedness) are the most common refractive errors, and the leading causes of visual impairment and blindness in the world. The goal of this study is to identify rare and low-frequency variants that influence spherical equivalent. Methods: We conducted variant-level and gene-level quantitative trait association analyses for mean spherical equiva- lent, using data from 1,560 individuals in the Beaver Dam Eye Study. Genotyping was conducted using the Illumina exome array. We analyzed 34,976 single nucleotide variants and 11,571 autosomal genes across the genome, using single-variant tests as well as gene-based tests. Results: Spherical equivalent was significantly associated with five genes in gene-based analysis: PTCHD2 at 1p36.22 (p = 3.6 × 10−7), CRISP3 at 6p12.3 (p = 4.3 × 10−6), NAP1L4 at 11p15.5 (p = 3.6 × 10−6), FSCB at 14q21.2 (p = 1.5 × 10−7), and AP3B2 at 15q25.2 (p = 1.6 × 10−7). -

The Detailed 3D Multi-Loop Aggregate/Rosette Chromatin Architecture and Functional Dynamic Organization of the Human and Mouse G
bioRxiv preprint doi: https://doi.org/10.1101/064642; this version posted August 15, 2016. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. The Detailed 3D Multi-Loop Aggregate/Rosette Chromatin Architecture and Functional Dynamic Organization of the Human and Mouse Genomes Tobias A. Knoch1*, Malte Wachsmuth2, Nick Kepper1,4, Michael Lesnussa1, Anis Abuseiris1, A. M. Ali Imam1,3, Petros Kolovos1,3, Jessica Zuin5, Christel E. M. Kockx6, Rutger W. W. Brouwer6, Harmen J. G. van de Werken3, Wilfred F. J. van IJken6, Kerstin S. Wendt5, and Frank G. Grosveld3 1Biophysical Genomics, Dept. Cell Biology & Genetics, Erasmus MC, Wytemaweg 80, 3015 CN Rotterdam, The Netherlands 2Cell Biology & Biophysics Unit, European Molecular Biology Laboratory, Meyerhofstr. 1, 69117 Heidelberg, Germany 3Cell Biology, Dept. Cell Biology & Genetics, Erasmus MC, Dr. Molewaterplein 50, 3015 GE Rotterdam, The Netherlands 4Genome Organization & Function, BioQuant & German Cancer Research Center, Im Neuenheimer Feld 267, 69120 Heidelberg, Germany 5Cohesin in Chromatin Structure and Gene Regulation, Dept. Cell Biology & Genetics, Erasmus MC, Dr. Molewaterplein 50, 3015 GE Rotterdam, The Netherlands 6Center for Biomics, Dept. Cell Biology & Genetics, Erasmus MC, Dr. Molewaterplein 50, 3015 GE Rotterdam, The Netherlands * corresponding author: [email protected] Knoch et al. 1 bioRxiv preprint doi: https://doi.org/10.1101/064642; this version posted August 15, 2016. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Abstract: The dynamic three-dimensional chromatin architecture of genomes and its co- evolutionary connection to its function – the storage, expression, and replication of genetic information – is still one of the central issues in biology. -

Transcriptional Regulators Are Upregulated in the Substantia Nigra
Journal of Emerging Investigators Transcriptional Regulators are Upregulated in the Substantia Nigra of Parkinson’s Disease Patients Marianne Cowherd1 and Inhan Lee2 1Community High School, Ann Arbor, MI 2miRcore, Ann Arbor, MI Summary neurological conditions is an established practice (3). Parkinson’s disease (PD) affects approximately 10 Significant gene expression dysregulation in the SN and million people worldwide with tremors, bradykinesia, in the striatum has been described, particularly decreased apathy, memory loss, and language issues. Though such expression in PD synapses. Protein degradation has symptoms are due to the loss of the substantia nigra (SN) been found to be upregulated (4). Mutations in SNCA brain region, the ultimate causes and complete pathology are unknown. To understand the global gene expression (5), LRRK2 (6), and GBA (6) have also been identified changes in SN, microarray expression data from the SN as familial markers of PD. SNCA encodes alpha- tissue of 9 controls and 16 PD patients were compared, synuclein, a protein found in presynaptic terminals that and significantly upregulated and downregulated may regulate vesicle presence and dopamine release. genes were identified. Among the upregulated genes, Eighteen SNCA mutations have been associated with a network of 33 interacting genes centered around the PD and, although the exact pathogenic mechanism is cAMP-response element binding protein (CREBBP) was not confirmed, mutated alpha-synuclein is the major found. The downstream effects of increased CREBBP- component of protein aggregates, called Lewy bodies, related transcription and the resulting protein levels that are often found in PD brains and may contribute may result in PD symptoms, making CREBBP a potential therapeutic target due to its central role in the interactive to cell death. -

Molecular Targeting and Enhancing Anticancer Efficacy of Oncolytic HSV-1 to Midkine Expressing Tumors
University of Cincinnati Date: 12/20/2010 I, Arturo R Maldonado , hereby submit this original work as part of the requirements for the degree of Doctor of Philosophy in Developmental Biology. It is entitled: Molecular Targeting and Enhancing Anticancer Efficacy of Oncolytic HSV-1 to Midkine Expressing Tumors Student's name: Arturo R Maldonado This work and its defense approved by: Committee chair: Jeffrey Whitsett Committee member: Timothy Crombleholme, MD Committee member: Dan Wiginton, PhD Committee member: Rhonda Cardin, PhD Committee member: Tim Cripe 1297 Last Printed:1/11/2011 Document Of Defense Form Molecular Targeting and Enhancing Anticancer Efficacy of Oncolytic HSV-1 to Midkine Expressing Tumors A dissertation submitted to the Graduate School of the University of Cincinnati College of Medicine in partial fulfillment of the requirements for the degree of DOCTORATE OF PHILOSOPHY (PH.D.) in the Division of Molecular & Developmental Biology 2010 By Arturo Rafael Maldonado B.A., University of Miami, Coral Gables, Florida June 1993 M.D., New Jersey Medical School, Newark, New Jersey June 1999 Committee Chair: Jeffrey A. Whitsett, M.D. Advisor: Timothy M. Crombleholme, M.D. Timothy P. Cripe, M.D. Ph.D. Dan Wiginton, Ph.D. Rhonda D. Cardin, Ph.D. ABSTRACT Since 1999, cancer has surpassed heart disease as the number one cause of death in the US for people under the age of 85. Malignant Peripheral Nerve Sheath Tumor (MPNST), a common malignancy in patients with Neurofibromatosis, and colorectal cancer are midkine- producing tumors with high mortality rates. In vitro and preclinical xenograft models of MPNST were utilized in this dissertation to study the role of midkine (MDK), a tumor-specific gene over- expressed in these tumors and to test the efficacy of a MDK-transcriptionally targeted oncolytic HSV-1 (oHSV). -

Dissertation Biochemical, Biophysical and Functional
DISSERTATION BIOCHEMICAL, BIOPHYSICAL AND FUNCTIONAL CHARACTERIZATION OF HISTONE CHAPERONES Submitted by Ling Zhang Department of Biochemistry and Molecular Biology In partial fulfillment of the requirements For the Degree of Doctor of Philosophy Colorado State University Fort Collins, Colorado Spring 2014 Doctoral committee: Advisor: Karolin Luger Diego Krapf Jennifer Nyborg Alan van Orden Laurie Stargell ABSTRACT BIOCHEMICAL, BIOPHYSICAL AND FUNCTIONAL CHARACTERIZATION OF HISTONE CHAPERONES Nucleosomes, the basic repeating unit of chromatin, are highly dynamic. Nucleosome dynamics allow for various cellular activities such as replication, recombination, transcription and DNA repair, while maintaining a high degree of DNA compaction. Each nucleosome is composed of 147 bp DNA wrapping around a histone octamer. Histone chaperones interact with histones and regulate nucleosome assembly and disassembly in the absence of ATP. To understand how nucleosome dynamics are regulated, it is essential to characterize the functions of histone chaperones. The first project of my doctoral research focused on the comparison of different nucleosome assembly proteins employing various biochemical and molecular approaches. Nucleosome assembly proteins (Nap) are a large family of histone chaperones, including Nap1 and Vps75 in Saccharomyces cerevisiae, and Nap1 (also Nap1L1), Nap1L2-6 (Nap1-like 2-6, with Nap1L4 being Nap2) and Set in metazoans. The functional differences of nucleosome assembly proteins are thus interesting to explore. We show that Nap1, Nap2 and Set bind to histones with similar and high affinities, but Nap2 and Set do not disassemble non-nucleosomal DNA-histone complexes as efficiently as Nap1. Also, nucleosome assembly proteins do not display discrepancies for histone variants or different DNA sequences. ii In the second project, we identified Spn1 as a novel histone chaperone and look into new functions of Spn1 on the regulation of chromatin structural states. -

Alternative Mechanisms Associated with Silencing of CDKN1C in Beckwith–Wiedemann Syndrome N Diaz-Meyer, Y Yang, S N Sait, E R Maher, M J Higgins
648 ORIGINAL ARTICLE J Med Genet: first published as 10.1136/jmg.2004.030593 on 1 August 2005. Downloaded from Alternative mechanisms associated with silencing of CDKN1C in Beckwith–Wiedemann syndrome N Diaz-Meyer, Y Yang, S N Sait, E R Maher, M J Higgins ............................................................................................................................... J Med Genet 2005;42:648–655. doi: 10.1136/jmg.2004.030593 Background: Mutations in the imprinted gene CDKN1C account for approximately 10% of Beckwith– Wiedemann syndrome (BWS) cases. Fibroblasts from BWS patients with loss of methylation (LOM) at the imprinting control region (ICR) KvDMR1 have reduced CDKN1C expression. Another group of BWS patients with downregulated CDKN1C expression but with normal methylation at KvDMR1 has been identified. See end of article for Objective: To investigate the mechanism of CDKN1C silencing in BWS in these two classes of patients. authors’ affiliations ....................... Methods: The CDKN1C promoter region was analysed for changes in DNA methylation using bisulphite sequencing, and for alterations in chromatin structure using the chromatin immunoprecipitation (ChIP) Correspondence to: assay. Michael J Higgins, Results: There was only spurious CpG methylation of the CDKN1C promoter in fibroblast DNA from both Department of Cancer Genetics, Elm and Carlton normal individuals and patients with BWS, irrespective of the methylation status of KvDMR1. There was no Streets, Roswell Park detectable change in chromatin structure at the CDKN1C promoter in patients with LOM at KvDMR1. BWS Cancer Institute, Buffalo, patients with downregulated CDKN1C and normal methylation at KvDMR1 had depletion of dimethylated NY 14263, USA; michael. [email protected] H3-K4 and enrichment of dimethylated H3-K9 and HP1c at the CDKN1C promoter, suggesting that in these cases gene silencing is associated with repressive chromatin changes.